
Reglas de Woodward–Hoffmann
Las reglas de Woodward-Hoffmann (o reglas de selección pericíclicas), ideadas por Robert Burns Woodward y Roald Hoffmann, son un conjunto de reglas utilizadas para racionalizar o predecir ciertos aspectos de la estereoquímica y la energía de activación de las reacciones pericíclicas, una clase importante de reacciones en química Orgánica. Las reglas se entienden mejor en términos del concepto de conservación de la simetría orbital utilizando diagramas de correlación orbital.(consulte la Sección 3 a continuación). Las reglas de Woodward-Hoffmann son una consecuencia de los cambios en la estructura electrónica que ocurren durante una reacción pericíclica y se basan en la fase de los orbitales moleculares que interactúan. Son aplicables a todas las clases de reacciones pericíclicas (y sus procesos 'retro' inversos microscópicos), incluidas (1) electrociclaciones, (2) cicloadiciones, (3) reacciones sigmatrópicas, (4) reacciones de transferencia de grupo, (5) reacciones de eno, (6) reacciones queletrópicas y (7) reacciones diotrópicas. Debido a su elegancia, simplicidad y generalidad, se atribuye a las reglas de Woodward-Hoffmann el primer ejemplo del poder de la teoría de orbitales moleculares para los químicos experimentales.
Woodward y Hoffmann desarrollaron las reglas de selección pericíclica al examinar las correlaciones entre los orbitales reactivo y producto (es decir, cómo los orbitales reactivo y producto se relacionan entre sí mediante distorsiones geométricas continuas que son funciones de la coordenada de reacción). Identificaron la conservación de la simetría orbital.como un principio teórico crucial que dicta el resultado (o la viabilidad) de un proceso pericíclico. También se han propuesto otros enfoques teóricos que conducen a las mismas reglas de selección. Hoffmann recibió el Premio Nobel de Química de 1981 por dilucidar la importancia de la simetría orbital en las reacciones pericíclicas, que compartió con Kenichi Fukui. Fukui desarrolló un conjunto similar de ideas en el marco de la teoría de los orbitales moleculares fronterizos (FMO). Debido a que Woodward había muerto dos años antes, no era elegible para ganar el que habría sido su segundo Premio Nobel de Química.
Antecedentes y terminología
Una reacción pericíclica es una reacción orgánica que procede a través de un solo estado de transición cíclico y concertado, cuya geometría permite la superposición continua de un ciclo de orbitales (π y/o σ). En el lenguaje de la simetría orbital, una reacción pericíclica se denomina simetría prohibida si existe una barrera energética adicional impuesta por la simetría que surge de la correlación prevista de la configuración electrónica del estado fundamental del material de partida con una configuración electrónica del estado excitado del producto y viceversa. (Aunque la regla de no cruce prohíbe tal correlación, el aumento de energía a medida que se acerca el cruce previsto da como resultado una barrera de energía adicional). Una reacción pericíclica se clasifica como simetría permitida.si no existe tal barrera impuesta por la simetría. Por lo tanto, estos términos no implican si la reacción en cuestión realmente tendrá lugar. Más bien, con todos los demás factores energéticos siendo iguales, un proceso prohibido por la simetría se verá obstaculizado por una barrera energética adicional. Aunque la barrera impuesta por la simetría suele ser formidable (hasta aprox. 5 eV o 480 kJ/mol en el caso de una cicloadición [2+2] prohibida), la prohibición no es absoluta y aún pueden tener lugar reacciones prohibidas por la simetría. a través de una vía pericíclica si otros factores (p. ej., liberación de tensión) favorecen la reacción. Del mismo modo, una reacción permitida por la simetría puede ser adelantada por una barrera energética infranqueable resultante de factores no relacionados con la simetría orbital.
Las reglas de Woodward-Hoffmann se formularon por primera vez en 1965 para explicar la sorprendente estereoespecificidad de las reacciones electrocíclicas bajo control térmico y fotoquímico. La estereoquímica de la electrociclación fue sintéticamente importante en el contexto del largo esfuerzo de Woodward por sintetizar la vitamina B 12, y las observaciones realizadas en el curso de la síntesis sirvieron para inspirar la formulación de las reglas de Woodward-Hoffmann. La interconversión de los derivados modelo de ciclobuteno y butadieno en condiciones térmicas (calentamiento) y fotoquímicas (irradiación ultravioleta) es ilustrativa. La termólisis de trans -1,2,3,4-tetrametil-1-ciclobuteno (1) proporcionó solo un isómero geométrico, (E, E)-3,4-dimetil-2,4-hexadieno (2); los isómeros geométricos (Z, Z) y (E, Z) no se detectaron en la mezcla del producto. De manera similar, la termólisis de cis -1,2,3,4-tetrametil-1-ciclobuteno (3) proporcionó solo el isómero 4 (E, Z). En ambas reacciones de apertura del anillo, los carbonos en los extremos del enlace σ que se rompe giran en la misma dirección. Por otro lado, se siguió el curso estereoquímico opuesto bajo activación fotoquímica: cuando el compuesto relacionado (E, E)-2,4-hexadieno (5) se expuso a la luz, se formó cis -3,4-dimetil-1-ciclobuteno (6) exclusivamente como resultado del cierre del anillo electrocíclico. Esto requiere que los extremos del sistema π giren en direcciones opuestas para formar el nuevo enlace σ. La termólisis de 6 sigue el mismo curso estereoquímico que 3: la apertura del anillo electrocíclico conduce a la formación de (E, Z)-2,4-hexadieno (7) y no de 5.
Los términos conrotatorio y disrotatorio se acuñaron para describir el sentido relativo de la rotación del enlace involucrado en las reacciones de apertura y cierre del anillo electrocíclico. Cuando los dos extremos del enlace que se rompe o forma giran en la misma dirección (ambos en el sentido de las agujas del reloj o en el sentido contrario a las agujas del reloj, como en el caso de la apertura del anillo de 1, 3 o 6 en condiciones térmicas), el proceso se denomina conrotatorio. Cuando los dos extremos giran en direcciones opuestas (uno en el sentido de las agujas del reloj y otro en el sentido contrario a las agujas del reloj, como en el cierre del anillo fotoquímico de 5), el proceso se denomina disrotatorio. Se encontró que 4Las reacciones electrocíclicas fotoquímicas de n - electrones y térmicas de (4 n + 2) electrones fueron conrotatorias en general, mientras que las reacciones fotoquímicas de 4 n - electrones y electrocíclicas térmicas de (4 n + 2) electrones fueron disrotatorias en general. Este patrón se explicó por primera vez en 1965, cuando Woodward y Hoffmann propusieron la conservación de la simetría orbital (ver más abajo) como un principio clave que gobierna el curso estereoquímico de las reacciones electrocíclicas.
Eventualmente, se reconoció que las reacciones pericíclicas promovidas térmicamente en general obedecen a un solo conjunto de reglas de selección generalizadas, dependiendo del conteo de electrones y la topología de las interacciones orbitales. El concepto clave de topología orbital o facialidad se introdujo para unificar varias clases de reacciones pericíclicas bajo un marco conceptual único. En resumen, un conjunto de átomos contiguos y sus orbitales asociados que reaccionan como una unidad en una reacción pericíclica se conoce como componente, y se dice que cada componente es antarafacial o suprafacial.dependiendo de si los lóbulos orbitales que interactúan durante la reacción están en el lado opuesto o en el mismo lado del plano nodal, respectivamente. (Los términos más antiguos conrotatorio y disrotatorio, que se aplican solo a la apertura y cierre de anillos electrocíclicos, se incluyen en los términos antarafacial y suprafacial, respectivamente, en este sistema de clasificación más general). Dadas estas definiciones generales, las reglas de Woodward-Hoffmann pueden ser dicho sucintamente en una sola oración:
Regla de selección pericíclica generalizada. Un proceso pericíclico de estado fundamental que involucra N pares de electrones y A componentes antarafaciales está permitido por simetría si y solo si N + A es impar.
Un proceso pericíclico de estado fundamental se produce mediante la adición de energía térmica (es decir, calentando el sistema, simbolizado por Δ). En contraste, un proceso pericíclico de estado excitado tiene lugar si un reactivo es promovido a un estado electrónicamente excitado por activación con luz ultravioleta (es decir, irradiando el sistema, simbolizado por hv). Sin embargo, es importante reconocer que el mecanismo operativo de una reacción formalmente pericíclica que tiene lugar bajo irradiación fotoquímica generalmente no es tan simple ni tan claro como sugiere esta dicotomía. Por lo general, son posibles varios modos de excitación electrónica, y las moléculas excitadas electrónicamente pueden cruzarse entre sistemas, decaer sin radiación o relajarse hasta una geometría de equilibrio desfavorable antes de que pueda tener lugar el proceso pericíclico del estado excitado. Por lo tanto, se cree que muchas reacciones pericíclicas aparentes que tienen lugar bajo la irradiación son procesos escalonados que involucran intermediarios dirradicalarios. Sin embargo, se observa con frecuencia que las reglas de selección pericíclica se invierten cuando se cambia de activación térmica a fotoquímica. Esto se puede racionalizar considerando la correlación de los primeros estados electrónicos excitados de los reactivos y productos. Aunque es más una heurística útil que una regla, se puede establecer un principio de selección generalizado correspondiente para las reacciones pericíclicas fotoquímicas:Un proceso pericíclico que involucra N pares de electrones y A componentes antarafaciales a menudo se ve favorecido en condiciones fotoquímicas si N + A es par. También se conocen reacciones pericíclicas que involucran un número impar de electrones. Con respecto a la aplicación de la regla de selección pericíclica generalizada, estos sistemas generalmente pueden tratarse como si estuviera involucrado un electrón más.
Entre el desarrollo inicial del principio de conservación de la simetría orbital en 1965 por Woodward y Hoffmann y su declaración de la regla de selección pericíclica generalizada en 1969, Howard Zimmerman y Michael JS Dewar propusieron un marco conceptual igualmente general, conocido como el concepto de Möbius-Hückel. o la teoría del estado de transición aromática para explicar la reactividad y selectividad de los sistemas pericíclicos, mientras que Kenichi Fukui analizó los sistemas pericíclicos utilizando los principios de la teoría orbital de frontera. En el enfoque de Dewar-Zimmerman, la topología de la superposición de orbitales (Hückel o Möbius) y el recuento total de electrones del sistema (4 n + 2 o 4 n) dan como resultado estados de transición que se clasifican como aromáticos o antiaromáticos. En el lenguaje de la teoría del estado de transición aromático, las reglas de Woodward-Hoffmann se pueden reformular de la siguiente manera: un estado de transición pericíclico que involucra (4 n + 2) electrones con topología de Hückel o 4 n electrones con topología de Möbius es aromático y está permitido, mientras que estado de transición que involucra 4 n -electrones con topología de Hückel o (4 n+ 2)-los electrones con topología de Möbius son antiaromáticos y prohibidos. El enfoque de Fukui, por otro lado, analiza las interacciones entre el HOMO y el LUMO de cada uno de los reactivos, o dentro de un reactivo. Un proceso en el que la interacción HOMO-LUMO es constructiva (resulta en una interacción de enlace neto) es favorable y se considera que permite la simetría, mientras que un proceso en el que la interacción HOMO-LUMO no es constructiva (resulta en interacciones de enlace y antienlace que cancelan) es desfavorable y se considera simetría prohibida. El enfoque del diagrama de correlación (la conservación de la simetría orbital, vide supra), propuesto por Woodward y Hoffmann y aclarado por Longuet-Higgins y otros, condujo a la afirmación general de que se permite una reacción pericíclica si la suma del número de suprafaciales 4q + 2 componentes y el número de componentes antarafaciales 4 r es impar. Es importante destacar que, aunque conceptualmente distintas, la teoría del estado de transición aromática (Zimmerman y Dewar), la teoría de la frontera molecular orbital (Fukui) y el principio de conservación de la simetría orbital (Woodward y Hoffmann) hacen predicciones idénticas.
Aunque la "simetría" orbital se utiliza como herramienta para dibujar diagramas de correlación orbital y de estado, la presencia o ausencia absoluta de un elemento de simetría no es fundamental para determinar si una reacción está permitida o prohibida. Es decir, la introducción de un sustituyente simple que interrumpe formalmente un plano o eje de simetría (p. ej., un grupo metilo) generalmente no afecta la evaluación de si una reacción está permitida o prohibida. En cambio, la simetría presente en un análogo no sustituido se usa para simplificar la construcción de diagramas de correlación orbital y evitar la necesidad de realizar cálculos. Solo las relaciones de fase entre los orbitales son importantes al juzgar si una reacción está permitida o prohibida por "simetría". Además, todavía se pueden hacer correlaciones orbitales, incluso si no hay elementos de simetría conservados (p. ej., desplazamientos 1,5-sigmatrópicos y reacciones ene). Por esta razón, los análisis de Woodward-Hoffmann, Fukui y Dewar-Zimmerman son igualmente amplios en su aplicabilidad, aunque cierto enfoque puede ser más fácil o más intuitivo de aplicar que otro, dependiendo de la reacción que se desee analizar.
Formulación original
Las reglas de Woodward-Hoffmann se invocaron por primera vez para explicar la estereoespecificidad observada de las reacciones electrocíclicas de apertura y cierre del anillo en los extremos de los polienos conjugados de cadena abierta, ya sea mediante la aplicación de calor (reacciones térmicas) o la aplicación de luz (reacciones fotoquímicas).
En la publicación original de 1965, las tres reglas extraídas de la evidencia experimental y el análisis orbital molecular aparecían de la siguiente manera:
- En un sistema de cadena abierta que contiene 4 n π electrones, la simetría orbital del orbital molecular ocupado más alto es tal que una interacción de enlace entre los extremos debe implicar la superposición entre las envolturas orbitales en las caras opuestas del sistema y esto solo se puede lograr en un proceso conrotatorio.
- En sistemas abiertos que contienen (4 n + 2) electrones π, la interacción de enlace terminal dentro de las moléculas del estado fundamental requiere la superposición de las envolturas orbitales en la misma cara del sistema, que solo se puede lograr mediante desplazamientos disrotatorios.
- En una reacción fotoquímica, un electrón en el HOMO del reactivo se promueve a un estado excitado que conduce a una inversión de las relaciones de simetría terminal y la estereoespecificidad.
Con esta formulación, es posible comprender la estereoespecificidad del cierre del anillo electrocíclico del buta-1,3-dieno sustituido que se muestra a continuación. El buta-1,3-dieno tiene 4 

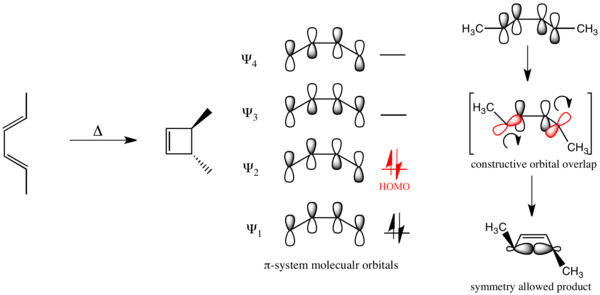
Por el contrario, en el cierre de anillo electrocíclico del hexa-1,3,5-trieno sustituido que se muestra a continuación, la reacción procede a través de un mecanismo disrotatorio.

En el caso de un cierre de anillo electrocíclico impulsado fotoquímicamente de buta-1,3-dieno, la promoción electrónica hace 
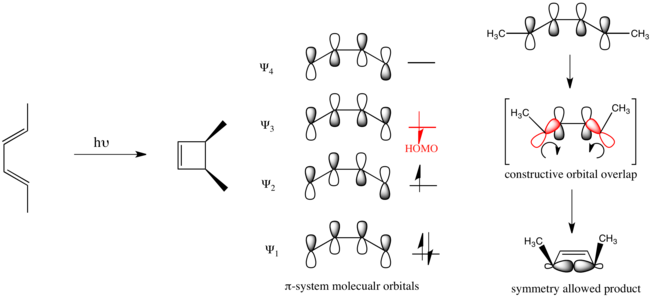
Se dice que las reacciones orgánicas que obedecen estas reglas tienen simetría permitida. Las reacciones que toman el curso opuesto están prohibidas por la simetría y requieren sustancialmente más energía para llevarse a cabo, si es que se llevan a cabo.
Diagramas de correlación
Como lo muestran Longuet-Higgins y EW Abrahamson, las reglas de Woodward-Hoffmann se pueden derivar mejor examinando el diagrama de correlación de una reacción dada. Un elemento de simetría es un punto de referencia (generalmente un plano o una línea) sobre el cual un objeto es simétrico con respecto a una operación de simetría. Si un elemento de simetría está presente durante todo el mecanismo de reacción (reactivo, estado de transición y producto), se denomina elemento de simetría conservada. Luego, a lo largo de la reacción, se debe conservar la simetría de los orbitales moleculares con respecto a este elemento. Es decir, los orbitales moleculares que son simétricos con respecto al elemento de simetría en el material de partida deben correlacionarse con (transformarse en) orbitales simétricos con respecto a ese elemento en el producto. Por el contrario, la misma afirmación se cumple para la antisimetría con respecto a un elemento de simetría conservado. Un diagrama de correlación de orbitales moleculares correlaciona los orbitales moleculares de los materiales de partida y el producto en función de la conservación de la simetría. A partir de unadiagrama de correlación de orbitales moleculares se puede construir un diagrama de correlación de estados electrónicos que correlacione los estados electrónicos (es decir, el estado fundamental y los estados excitados) de los reactivos con los estados electrónicos de los productos. Los diagramas de correlación se pueden usar para predecir la altura de las barreras del estado de transición.
Reacciones electrocíclicas
Teniendo en cuenta el cierre del anillo electrocíclico del 1,3-butadieno sustituido, la reacción puede proceder a través de un mecanismo de reacción conrotatorio o disrotatorio. Como se muestra a la izquierda, en el estado de transición conrotatorio hay un eje de simetría C 2 y en el estado de transición disrotatorio hay un plano de simetría especular σ. Para correlacionar los orbitales del material de partida y el producto, se debe determinar si los orbitales moleculares son simétricos o antisimétricos con respecto a estos elementos de simetría. Los orbitales moleculares del sistema π del butadieno se muestran a la derecha junto con el elemento de simetría con el que son simétricos. Son antisimétricos con respecto al otro. Por ejemplo, Ψ 2 de 1,3-butadieno es simétrico con respecto a 180rotación sobre el eje C 2 y antisimétrica con respecto a la reflexión en el plano del espejo.
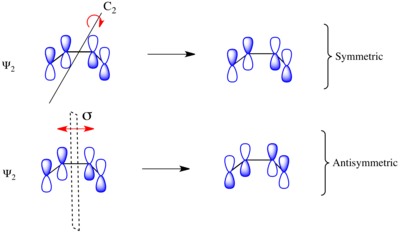
Ψ 1 y Ψ 3 son simétricos con respecto al plano del espejo ya que el signo de los lóbulos del orbital p se conserva bajo la transformación de simetría. De manera similar, Ψ 1 y Ψ 3 son antisimétricos con respecto al eje C 2 ya que la rotación invierte el signo de los lóbulos orbitales p uniformemente. Por el contrario, Ψ 2 y Ψ 4 son simétricos con respecto al eje C 2 y antisimétricos con respecto al plano del espejo σ.

El mismo análisis se puede realizar para los orbitales moleculares del ciclobuteno. El resultado de ambas operaciones de simetría en cada uno de los MO se muestra a la izquierda. Como los orbitales σ y σ se encuentran completamente en el plano que contiene C 2 perpendicular a σ, son uniformemente simétricos y antisimétricos (respectivamente) para ambos elementos de simetría. Por otro lado, π es simétrico respecto a la reflexión y antisimétrico respecto a la rotación, mientras que π es antisimétrico respecto a la reflexión y simétrico respecto a la rotación.
Las líneas de correlación se dibujan para conectar los orbitales moleculares en el material de partida y el producto que tienen la misma simetría con respecto al elemento de simetría conservada. En el caso del cierre del anillo electrocíclico conrotatorio de 4 electrones del 1,3-butadieno, el orbital molecular más bajo Ψ 1 es asimétrico (A) con respecto al eje C 2. Entonces, este orbital molecular está correlacionado con el orbital π del ciclobuteno, el orbital de menor energía que también es (A) con respecto al eje C 2. De manera similar, Ψ 2, que es simétrico (S) con respecto al eje C 2, está correlacionado con σ del ciclobuteno. Las dos correlaciones finales son entre los orbitales moleculares antisimétricos (A) Ψ 3 y σ, y los orbitales moleculares simétricos (S) Ψ 4 y π.

De manera similar, existe un diagrama de correlación para un mecanismo disrotatorio. En este mecanismo, el elemento de simetría que persiste a lo largo de todo el mecanismo es el plano de reflexión del espejo σ. Aquí, el MO Ψ 1 de energía más baja del 1,3-butadieno es simétrico con respecto al plano de reflexión y, como tal, se correlaciona con el MO simétrico σ del ciclobuteno. De manera similar, el par de energía más alta de orbitales moleculares simétricos Ψ 3 y π se correlacionan. En cuanto a los orbitales moleculares asimétricos, el par de menor energía Ψ 2 y π forman un par de correlación, al igual que Ψ 4 y σ.

Al evaluar los dos mecanismos, se predice que el mecanismo conrotatorio tiene una barrera más baja porque transforma los electrones de los orbitales en estado fundamental de los reactivos (Ψ 1 y Ψ 2) en orbitales en estado fundamental del producto (σ y π). Por el contrario, el mecanismo disrotatorio fuerza la conversión del orbital Ψ 1 en el orbital σ, y el orbital Ψ 2 en el orbital π. Así, los dos electrones en el estado fundamental del orbital Ψ 2 se transfieren a un orbital antienlazante excitado, creando un estado electrónico doblemente excitado del ciclobuteno. Esto conduciría a una barrera del estado de transición significativamente más alta para la reacción.
Sin embargo, como las reacciones no tienen lugar entre orbitales moleculares separados, sino entre estados electrónicos, el análisis final implica diagramas de correlación de estados. Un diagrama de correlación de estado correlaciona la simetría general de los estados electrónicos en el material de partida y el producto. El estado fundamental del 1,3-butadieno, como se muestra arriba, tiene 2 electrones en Ψ 1 y 2 electrones en Ψ 2, por lo que se representa como Ψ 1 Ψ 2. La simetría general del estado es el producto de las simetrías de cada orbital lleno con multiplicidad para orbitales doblemente poblados. Así, como Ψ 1 es asimétrica con respecto al eje C 2, y Ψ 2 es simétrica, el estado total está representado por As _ Para ver por qué este producto en particular es matemáticamente S total, ese S se puede representar como (+1) y A como (−1). Esto se deriva del hecho de que los signos de los lóbulos de los orbitales p se multiplican por (+1) si son simétricos con respecto a una transformación de simetría (es decir, inalterados) y se multiplican por (−1) si son antisimétricos con respecto a una transformación de simetría (es decir, invertida). Así A S =(−1) (+1) =+1=S. El primer estado excitado (ES-1) se forma a partir de la promoción de un electrón del HOMO al LUMO y, por lo tanto, se representa como Ψ 1 Ψ 2 Ψ 3. Como Ψ 1 es A, Ψ 2 es S y Ψ 3es A, la simetría de este estado viene dada por A SA=A.
Ahora, considerando los estados electrónicos del producto, el ciclobuteno, el estado fundamental viene dado por σ π, que tiene simetría S A =S. El primer estado excitado (ES-1') se forma nuevamente a partir de una promoción de un electrón del HOMO al LUMO, por lo que en este caso se representa como σ ππ. La simetría de este estado es S AS=A.
El estado fundamental Ψ 1 Ψ 2 del 1,3-butadieno se correlaciona con el estado fundamental σ π del ciclobuteno, como se demuestra en el diagrama de correlación MO anterior. Ψ 1 se correlaciona con π y Ψ 2 se correlaciona con σ. Por lo tanto, los orbitales que forman Ψ 1 Ψ 2 deben transformarse en los orbitales que forman σ π bajo un mecanismo conrotatorio. Sin embargo, el estado ES-1 no se correlaciona con el estado ES-1' ya que los orbitales moleculares no se transforman entre sí bajo el requisito de simetría visto en el diagrama de correlación de orbitales moleculares. En cambio, como Ψ 1 se correlaciona con π, Ψ 2 se correlaciona con σ y Ψ 3se correlaciona con σ, el estado Ψ 1 Ψ 2 Ψ 3 intenta transformarse en π σσ, que es un estado excitado diferente. Entonces ES-1 intenta correlacionarse con ES-2'=σπ σ, que tiene una energía más alta que Es-1'. Similarmente ES-1'=σ ππ intenta correlacionar con ES-2=Ψ 1 Ψ 2 Ψ 4. Estas correlaciones en realidad no pueden tener lugar debido a la regla de la mecánica cuántica conocida como regla de cruce evitado. Esto dice que las configuraciones energéticas de la misma simetría no pueden cruzarse en un diagrama de correlación de niveles de energía. En resumen, esto es causado por la mezcla de estados de la misma simetría cuando se acercan lo suficiente en energía. Entonces, en cambio, se forma una barrera energética alta entre una transformación forzada de ES-1 en ES-1 '. En el siguiente diagrama, las correlaciones preferidas por simetría se muestran en líneas discontinuas y las líneas curvas en negrita indican la correlación real con la barrera energética alta.

El mismo análisis se puede aplicar al mecanismo disrotatorio para crear el siguiente diagrama de correlación de estado.

Por lo tanto, si la molécula está en el estado fundamental, procederá a través del mecanismo conrotatorio (es decir, bajo control térmico) para evitar una barrera electrónica. Sin embargo, si la molécula está en el primer estado excitado (es decir, bajo control fotoquímico), la barrera electrónica está presente en el mecanismo conrotatorio y la reacción procederá a través del mecanismo disrotatorio. Estos no son completamente distintos ya que tanto el mecanismo conrotatorio como el disrotatorio se encuentran en la misma superficie potencial. Por lo tanto, una afirmación más correcta es que a medida que una molécula en estado fundamental explora la superficie de energía potencial, es más probable que alcance la barrera de activación para experimentar un mecanismo conrotatorio.
Reacciones de cicloadición
Las reglas de Woodward-Hoffmann también pueden explicar las reacciones de cicloadición bimolecular a través de diagramas de correlación. Una cicloadición [ π p + π q ] reúne dos componentes, uno con p π-electrones y el otro con q π-electrones. Las reacciones de cicloadición se caracterizan además como suprafaciales (s) o antarafaciales (a) con respecto a cada uno de los componentes π. (Consulte a continuación "Formulación general" para obtener una descripción detallada de la generalización de la notación WH a todos los procesos pericíclicos).
[2+2] Cicloadiciones
Hay 4 posibles consecuencias estereoquímicas para la cicloadición [2+2]: [ π 2 s + π 2 s ], [ π 2 a + π 2 s ], [ π 2 s + π 2 a ], [ π 2 a + π2a ]. _ Se demostrará que el modo geométricamente más plausible [ π 2 s + π 2 s ] está prohibido en condiciones térmicas, mientras que el [ π 2 a + π2 s ], los enfoques [ π 2 s + π 2 a ] están permitidos desde el punto de vista de la simetría, pero son raros debido a una deformación desfavorable y un perfil estérico. Por otro lado, son comunes las cicloadiciones [2+2] bajo activación fotoquímica.
Considerando la cicloadición [ π 2 s + π 2 s ] Este mecanismo conduce a una retención de la estereoquímica en el producto, como se ilustra a la derecha. Hay dos elementos de simetría presentes en los materiales de partida, el estado de transición y el producto: σ 1 y σ 2. σ 1 es el plano especular entre las componentes perpendiculares a los orbitales p; σ 2 divide las moléculas por la mitad perpendiculares a los enlaces σ. Ambos son elementos de simetría local en el caso de que los componentes no sean idénticos.

Para determinar la simetría y la asimetría con respecto a σ 1 y σ 2, los orbitales moleculares del material de partida deben considerarse en tándem. La figura de la derecha muestra el diagrama de correlación de orbitales moleculares para la cicloadición [ π 2 s + π 2 s ]. Los dos orbitales moleculares π y π de los materiales de partida se caracterizan por su simetría con respecto primero a σ 1 y luego a σ 2. Del mismo modo, el σ y σLos orbitales moleculares del producto se caracterizan por su simetría. En el diagrama de correlación, las transformaciones de los orbitales moleculares en el transcurso de la reacción deben conservar la simetría de los orbitales moleculares. Así, π SS se correlaciona con σ SS, π AS se correlaciona con σ AS, π SA se correlaciona con σ SA, y finalmente π AA se correlaciona con σ AA. Debido a la conservación de la simetría orbital, el orbital enlazante π AS se ve obligado a correlacionarse con el orbital antienlazante σ AS. Por lo tanto, se pronostica una barrera alta.
Esto se precisa en el siguiente diagrama de correlación de estados. El estado fundamental en los materiales de partida es el estado electrónico donde π SS y π AS están doblemente poblados, es decir, el estado (SS) (AS). Como tal, este estado intenta correlacionarse con el estado electrónico en el producto donde tanto σ SS como σ AS están doblemente poblados, es decir, el estado (SS) (AS). Sin embargo, este estado no es ni el estado fundamental (SS) (SA) del ciclobutano, ni el primer estado excitado ES-1'=(SS) (SA)(AS), donde un electrón es promovido de HOMO a LUMO. Entonces, el estado fundamental de los reactivos intenta correlacionarse con un segundo estado excitado ES-2'=(SS)(AS).
De manera similar, el estado fundamental del producto ciclobutano, como se puede ver en el diagrama de orbitales moleculares anterior, es el estado electrónico en el que tanto σ SS como σ SA están doblemente poblados, es decir, el estado (SS) (SA). Esto intenta correlacionarse con el estado en el que π SS y π SA están doblemente poblados, es decir, un segundo estado excitado ES-2=(SS) (SA).

Finalmente, el primer estado excitado de los materiales de partida es la configuración electrónica donde π SS está doblemente poblado, y π AS y π SA están ocupados individualmente, es decir, el estado (SS) (AS)(SA). El primer estado excitado del producto es también el estado (SS) (SA)(AS) ya que σ SS está doblemente poblado y σ SA y σ AS están ocupados por separado. Así, estos dos estados excitados se correlacionan.
El estado fundamental de los materiales de partida solo intenta correlacionarse con el segundo estado excitado, ya que se evita un cruce en el medio debido a que los estados poseen la misma simetría general. Así, en realidad, el estado fundamental de los reactivos se transforma en el estado fundamental de los productos solo después de lograr una alta barrera energética. Sin embargo, no existe una gran barrera de activación si los reactivos se encuentran en el primer estado excitado. Por lo tanto, esta reacción procede fácilmente bajo control fotoquímico, pero tiene una barrera muy alta para la reacción bajo control térmico.
[4+2] cicloadiciones
Una cicloadición [4+2] es una reacción pericíclica concertada de 2 componentes ejemplificada por la reacción de Diels-Alder. El caso más simple es la reacción de 1,3-butadieno con etileno para formar ciclohexeno que se muestra a la izquierda.
Solo hay un elemento de simetría conservado en esta transformación: el plano del espejo que pasa por el centro de los reactivos, como se muestra a la izquierda. A partir de esto podemos asignar la simetría de los orbitales moleculares de los reactivos de manera muy sencilla. Los orbitales moleculares de los reactivos son simplemente el conjunto {Ψ 1, Ψ 2, Ψ 3, Ψ 4 } de orbitales moleculares de 1,3-butadieno que se muestra arriba, junto con π y π de etileno. Ψ 1 es simétrico, Ψ 2 es antisimétrico, Ψ 3 es simétrico y Ψ 4 es antisimétrico con respecto al plano del espejo. De manera similar, π es simétrico y π es antisimétrico con respecto al plano del espejo.

Los orbitales moleculares del producto son las combinaciones simétricas y antisimétricas de los dos enlaces σ y σ recién formados y los enlaces π y π como se muestra a continuación.
La correlación de los pares de orbitales en los materiales iniciales y el producto de la misma simetría y el aumento de energía da el diagrama de correlación a la derecha. Como esto transforma los orbitales moleculares de enlace del estado fundamental de los materiales de partida en los orbitales de enlace del estado fundamental del producto de una manera conservadora de simetría, se predice que no tendrá la gran barrera energética presente en la reacción del estado fundamental [2+2] anterior.
Para hacer el análisis preciso, se puede construir el diagrama de correlación de estados para la cicloadición general [4+2]. Como antes, el estado fundamental es el estado electrónico representado en el diagrama de correlación de orbitales moleculares de la derecha. Esto se puede describir como Ψ 1 π Ψ 2, de simetría total S S A =S. Esto se correlaciona con el estado fundamental del ciclohexeno σ S σ A π que también es S S A =S. Como tal, no se prevé que esta reacción del estado fundamental tenga una barrera impuesta por una alta simetría.
También se pueden construir las correlaciones de estado excitado como se hizo anteriormente. Aquí, hay una barrera energética alta para una reacción de Diels-Alder fotoinducida bajo una topología de enlace suprafacial-suprafacial debido al cruce evitado que se muestra a continuación.

Reacciones de transferencia de grupo
Las alturas de barrera impuestas por la simetría de las reacciones de transferencia de grupo también se pueden analizar mediante diagramas de correlación. Una reacción modelo es la transferencia de un par de átomos de hidrógeno del etano al perdeuterioetileno que se muestra a la derecha.
El único elemento de simetría conservado en esta reacción es el plano del espejo que pasa por el centro de las moléculas, como se muestra a la izquierda.
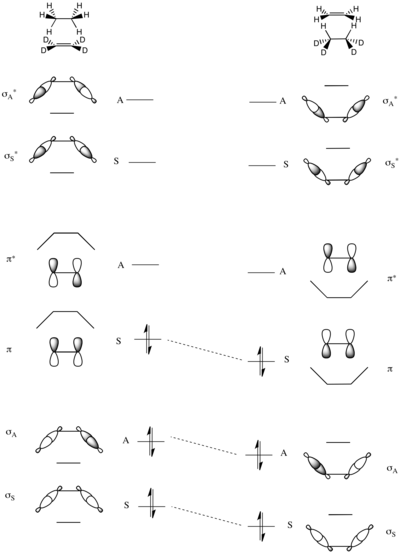
Los orbitales moleculares del sistema se construyen como combinaciones simétricas y antisimétricas de enlaces C-H σ y σ en el etano y enlaces π y π en el eteno deutero-sustituido. Por lo tanto, el OM de menor energía es la suma simétrica de los dos enlaces σ C–H (σ S), seguida de la suma antisimétrica (σ A). Los dos MO de mayor energía se forman a partir de combinaciones lineales de los antienlaces σ CH; el más alto es el σ A antisimétrico, precedido por el σ A simétrico con una energía ligeramente menor. En el medio de la escala energética están los dos OM restantes que son el π CC y el π CC del eteno.
El diagrama completo de correlación de orbitales moleculares se construye haciendo coincidir pares de OM simétricos y asimétricos de energía total creciente, como se explicó anteriormente. Como se puede ver en el diagrama adyacente, como los orbitales de enlace de los reactivos se correlacionan exactamente con los orbitales de enlace de los productos, no se prevé que esta reacción tenga una barrera impuesta por simetría electrónica alta.
Reglas de selección
Usando diagramas de correlación, se pueden derivar reglas de selección para las siguientes clases generalizadas de reacciones pericíclicas. Cada una de estas clases particulares se generaliza aún más en las reglas generalizadas de Woodward-Hoffmann. Los descriptores de topología de enlace más inclusivos antarafacial y suprafacial subsumen los términos conrotatorio y disrotatorio, respectivamente. Antarafacial se refiere a la formación o ruptura de enlaces a través de la cara opuesta de un sistema π, orbital p o enlace σ, mientras que suprafacial se refiere al proceso que ocurre a través de la misma cara. Una transformación suprafacial en un centro quiral conserva la estereoquímica, mientras que una transformación antarafacial invierte la estereoquímica.
Reacciones electrocíclicas
La regla de selección de las reacciones de electrociclación se da en el enunciado original de las reglas de Woodward-Hoffmann. Si se produce un cierre de anillo electrocíclico generalizado en un polieno de 4 n π-electrones, entonces es conrotatorio en condiciones térmicas y disrotatorio en condiciones fotoquímicas. Por el contrario, en un polieno de 4 n + 2 π-electrones, un cierre de anillo electrocíclico es disrotatorio en condiciones térmicas y conrotatorio en condiciones fotoquímicas.
Este resultado puede derivarse mediante un análisis FMO basado en el signo de los lóbulos orbitales p del HOMO del polieno o con diagramas de correlación. Tomando primero la primera posibilidad, en el estado fundamental, si un polieno tiene 4 n electrones, los orbitales p externos del HOMO que forman el enlace σ en el producto electrociclado son de signos opuestos. Así, una superposición constructiva sólo se produce bajo un proceso conrotatorio o antarafacial. Por el contrario, para un polieno con 4 n + 2 electrones, los orbitales p exteriores del estado fundamental HOMO son del mismo signo. Por lo tanto, la superposición orbital constructiva ocurre con un proceso disrotatorio o suprafacial.
Además, el diagrama de correlación para cualquier reacción electrocíclica de 4 n se parecerá al diagrama de la ciclación de 4 electrones del 1,3-butadieno, mientras que el diagrama de correlación de cualquier reacción electrocíclica de 4 n + 2 electrones se parecerá al diagrama de correlación de la ciclación de 6 electrones de 1,3,5-hexatrieno.
Esto se resume en la siguiente tabla:
| Térmicamente permitido | Permitido fotoquímicamente | |
|---|---|---|
| 4 norte | conrotatorio | disrotatorio |
| 4 n + 2 | disrotatorio | conrotatorio |
Reacciones de reordenamiento sigmatrópico
Un reordenamiento sigmatrópico general se puede clasificar como orden [ i, j ], lo que significa que un enlace σ originalmente entre los átomos indicados como 1 y 1', adyacentes a uno o más sistemas π, se desplaza entre los átomos i y j. Por lo tanto, migra (i − 1), (j − 1) átomos lejos de su posición original.
Un análisis de simetría formal a través de diagramas de correlación no es útil en el estudio de reordenamientos sigmatrópicos ya que, en general, solo hay elementos de simetría presentes en el estado de transición. Excepto en casos especiales (por ejemplo, reordenamientos [3,3]), no hay elementos de simetría que se conserven cuando se atraviesa la coordenada de reacción. Sin embargo, las correlaciones orbitales entre los materiales y productos de partida aún pueden analizarse, y las correlaciones de los orbitales de materiales de partida con los orbitales de productos de alta energía, como de costumbre, darán como resultado procesos de "simetría prohibida". Sin embargo, un enfoque basado en FMO (o el análisis de Dewar-Zimmerman) es más sencillo de aplicar.
Una de las clases más frecuentes de cambios sigmatrópicos se clasifica como [1, j ], donde j es impar. Eso significa que un extremo del enlace σ migra (j − 1) enlaces a través de un sistema π mientras que el otro extremo no migra. Es una reacción que involucra j + 1 electrones: j − 1 del sistema π y 2 del enlace σ. Usando el análisis FMO, se permiten reordenamientos [1, j ]-sigmatrópicos si el estado de transición tiene una superposición constructiva entre el grupo migratorio y el orbital p de aceptación del HOMO. En reordenamientos [1, j ]-sigmatrópicos si j + 1 = 4 n, entonces se permite térmicamente supra/antara, y si j + 1 = 4n + 2, entonces se permite térmicamente supra/supra o antara/antara.
La otra clase prevalente de reordenamientos sigmatrópicos son [3,3], en particular los reordenamientos de Cope y Claisen. Aquí, las interacciones constructivas deben darse entre los HOMO de los dos fragmentos de radicales alilo en el estado de transición. El estado fundamental HOMO Ψ 2 del fragmento de alilo se muestra a continuación. Como los orbitales p terminales son de signo opuesto, esta reacción puede tener lugar en una topología supra/supra o en una topología antara/antara.
Las reglas de selección para un reordenamiento [ i, j ]-sigmatrópico son las siguientes:
- Para supra/supra o antara/antara [ i, j ]-desplazamientos sigmatrópicos, si i + j = 4 n + 2 están permitidos térmicamente y si i + j = 4 n están permitidos fotoquímicamente
- Para desplazamientos supra/antara [ i, j ]-sigmatrópicos, si i + j = 4 n están permitidos térmicamente, y si i + j = 4 n + 2 están permitidos fotoquímicamente
Esto se resume en la siguiente tabla:
| yo + j | Térmicamente permitido | Permitido fotoquímicamente |
|---|---|---|
| 4 norte | yo s + j a o yo a + j s | yo s + j s o yo un + j un |
| 4 n + 2 | yo s + j s o yo un + j un | yo s + j a o yo a + j s |
Reacciones de cicloadición
Una cicloadición general [ p + q ] es una reacción de adición concertada entre dos componentes, uno con p electrones π y otro con q electrones π. Esta reacción es simétrica permitida bajo las siguientes condiciones:
- Para una cicloadición supra/supra o antara/antara, se permite térmicamente si p + q = 4 n + 2 y se permite fotoquímicamente si p + q = 4 n
- Para una cicloadición supra/antara, se permite térmicamente si p + q = 4 n y se permite fotoquímicamente si p + q = 4 n + 2
Esto se resume en la siguiente tabla:
| pag + q | Térmicamente permitido | Permitido fotoquímicamente |
|---|---|---|
| 4 norte | pags + qa o pags + qs _ _ _ _ | p s + q s o p a + q a |
| 4 n + 2 | p s + q s o p a + q a | pags + qa o pags + qs _ _ _ _ |
Reacciones de transferencia de grupo
Una reacción general de transferencia de doble grupo que es síncrona se puede representar como una interacción entre un componente con p π electrones y un componente con q π electrones, como se muestra.
Entonces las reglas de selección son las mismas que para las reacciones de cicloadición generalizadas. Eso es
- Para transferencias de doble grupo supra/supra o antara/antara, si p + q = 4 n + 2 se permite térmicamente, y si p + q = 4 n se permite fotoquímicamente
- Para transferencias de doble grupo supra/antara, si p + q = 4 n se permite térmicamente, y si p + q = 4 n + 2 se permite fotoquímicamente
Esto se resume en la siguiente tabla:
| pag + q | Térmicamente permitido | Permitido fotoquímicamente |
|---|---|---|
| 4 norte | pags + qa o pags + qs _ _ _ _ | p s + q s o p a + q a |
| 4 n + 2 | p s + q s o p a + q a | pags + qa o pags + qs _ _ _ _ |
El caso de q = 0 corresponde a la eliminación térmica de los grupos R "transferidos". Existe evidencia de que las eliminaciones pirolíticas de dihidrógeno y etano de 1,4-ciclohexadieno y 3,3,6,6-tetrametil-1,4-ciclohexadieno, respectivamente, representan ejemplos de este tipo de proceso pericíclico.
La reacción ene a menudo se clasifica como un tipo de proceso de transferencia de grupo, aunque no implica la transferencia de dos grupos con enlaces σ. Más bien, solo se transfiere un enlace σ mientras que un segundo enlace σ se forma a partir de un enlace π roto. Como un proceso completamente suprafacial que involucra 6 electrones, está permitido por simetría en condiciones térmicas. El símbolo de Woodward-Hoffmann para la reacción ene es [ π 2 s + π 2 s + σ 2 s ] (ver más abajo).
Formulación general
Aunque las reglas de Woodward-Hoffmann se establecieron por primera vez en términos de procesos electrocíclicos, eventualmente se generalizaron a todas las reacciones pericíclicas, como deberían indicar la similitud y los patrones en las reglas de selección anteriores.
En las reglas generalizadas de Woodward-Hoffmann, todo se caracteriza en términos de topologías de enlace antarafacial y suprafacial. Los términos conrotatorio y disrotatorio son suficientes para describir el sentido relativo de la rotación del enlace en las reacciones de cierre o apertura de anillos electrocíclicos, como se ilustra a la derecha. Sin embargo, no son adecuados para describir las topologías de formación y ruptura de enlaces que tienen lugar en una reacción pericíclica general. Como se describe en detalle a continuación, en la formulación general de las reglas de Woodward-Hoffmann, los términos de rotación de enlace conrotatorio y disrotatorio están subsumidos por los términos de topología de enlace (o facialidad) antarafacial y suprafacial, respectivamente. Estos descriptores se pueden utilizar para caracterizar la topología de la formación y ruptura de enlaces que tiene lugar en cualquier proceso pericíclico.
Notación de Woodward-Hoffmann
Un componente es cualquier parte de una molécula o moléculas que funcionan como una unidad en una reacción pericíclica. Un componente consta de uno o más átomos y cualquiera de los siguientes tipos de orbitales asociados:
- Un orbital p o sp aislado ( lleno o vacío, símbolo ω)
- Un sistema π conjugado (símbolo π)
- Un enlace σ (símbolo σ)
El recuento de electrones de un componente es el número de electrones en los orbitales del componente:
- El recuento de electrones de un orbital ω vacío (es decir, un orbital p vacío) es 0, mientras que el de un orbital ω lleno (es decir, un par solitario) es 2.
- El recuento de electrones de un sistema π conjugado con n dobles enlaces es 2 n (o 2 n + 2, si se conjuga un par solitario (formal) de un heteroátomo o carbanión).
- El conteo de electrones de un enlace σ es 2.
La topología de enlace de un componente puede ser suprafacial y antarafacial:
- La relación es suprafacial (símbolo: s) cuando las interacciones con el sistema π o el orbital p ocurren en el mismo lado del plano nodal (piense en syn). Para un enlace σ, corresponde a interacciones que ocurren en los dos lóbulos "interiores" o dos lóbulos "exteriores" del enlace.
- La relación es antarafacial (símbolo: a) cuando las interacciones con el sistema π o el orbital p ocurren en lados opuestos del plano nodal (piense en anti). Para un enlace σ, corresponde a interacciones que ocurren en un lóbulo "interior" y un lóbulo "exterior" del enlace.
Usando esta notación, a todas las reacciones pericíclicas se les puede asignar un descriptor, que consiste en una serie de símbolos σ/π/ω N s/a, conectados por signos + y encerrados entre paréntesis, que describen, en orden, el tipo de orbital(es), número de electrones y topología de enlace involucrada para cada componente. A continuación se muestran algunos ejemplos ilustrativos:
- La reacción de Diels-Alder (una cicloadición (4+2)) es [ π 4 s + π 2 s ].
- La cicloadición dipolar 1,3 de ozono y una olefina en el primer paso de la ozonólisis (una cicloadición (3+2)) es [ π 4 s + π 2 s ].
- La adición queletrópica de dióxido de azufre al 1,3-butadieno (una adición queletrópica (4+1)) es [ ω 0 a + π 4 s ] + [ ω 2 s + π 4 s ].
- El reordenamiento de Cope (un cambio [3,3]-sigmatrópico) es [ π 2 s + σ 2 s + π 2 s ] o [ π 2 a + σ 2 s + π 2 a ].
- La migración de [1,3]-alquilo con inversión en el carbono descubierta por Berson (un desplazamiento [1,3]-sigmatrópico) es [ σ 2 a + π 2 s ].
- El cierre del anillo electrocíclico conrotatorio del 1,3-butadieno (una electrociclación 4π) es [ π 4 a ].
- La apertura del anillo electrocíclico conrotatorio del ciclobuteno (una electrociclación 4π inversa) es [ σ 2 a + π 2 s ] o [ σ 2 s + π 2 a ].
- El cierre del anillo electrocíclico disrotatorio del anión 1,3-ciclooctadien-5-uro (una electrociclación 6π) es [ π 6 s ].
- Un desplazamiento Wagner-Meerwein de un carbocatión (un desplazamiento [1,2]-sigmatrópico) es [ ω 0 s + σ 2 s ].
Antarafacial y suprafacial están asociados con (conrotación o inversión) y (disrotación o retención), respectivamente. Un solo descriptor puede corresponder a dos procesos pericíclicos que son químicamente distintos, que una reacción y su reverso microscópico a menudo se describen con dos descriptores diferentes, y que un solo proceso puede tener más de un descriptor correcto. Se puede verificar, usando la regla de selección pericíclica dada a continuación, que todas estas reacciones son procesos permitidos.
Declaración original
Usando esta notación, Woodward y Hoffmann establecen en su revisión de 1969 la formulación general para todas las reacciones pericíclicas de la siguiente manera:
Se permite un cambio pericíclico del estado fundamental cuando el número total de componentes (4q+2) s y (4r) a es impar.
Aquí, (4 q + 2) s y (4 r) a se refieren a componentes suprafaciales (4 q + 2) de electrones y antarafaciales (4 r) de electrones, respectivamente. Además, este criterio debe interpretarse tanto como suficiente (indicado anteriormente) como necesario (no indicado explícitamente anteriormente, ver: si y solo si)
Derivación de un enunciado alternativo
Alternativamente, la declaración general se puede formular en términos del número total de electrones utilizando reglas simples de divisibilidad mediante un análisis directo de dos casos.
Primero, considere el caso donde el número total de electrones es 4 n + 2. Entonces podemos escribir4 norte + 2 = un (4 q + 2) s + segundo (4 pag + 2) un + c ( 4 t) s + re (4 r) un,
donde a, b, c y d son coeficientes que indican el número de cada tipo de componente. Esta ecuación implica que uno de, pero no ambos, a o b es impar, porque si a y b son ambos pares o ambos impares, entonces la suma de los cuatro términos es 0 (mod 4).
El enunciado generalizado de las reglas de Woodward-Hoffmann establece que a + d es impar si se permite la reacción. Ahora, si a es par, entonces esto implica que d es impar. Dado que b es impar en este caso, el número de componentes antarafaciales, b + d, es par. Asimismo, si a es impar, entonces d es par. Dado que b es par en este caso, el número de componentes antarafaciales, b + d, vuelve a ser par. Por lo tanto, independientemente de la suposición inicial de paridad para a y b, el número de componentes antarafaciales es par cuando el conteo de electrones es 4 n + 2. Por el contrario, en el caso prohibido, donde a + d es par, podemos demostrar que, independientemente de las suposiciones iniciales, b + d es impar.
En el caso en que el número total de electrones sea 4 n, argumentos similares (omitidos aquí) nos llevan a la conclusión de que el número de componentes antarafaciales b + d debe ser impar en el caso permitido e incluso en el caso prohibido.
Finalmente, para completar el argumento y mostrar que este nuevo criterio es verdaderamente equivalente al criterio original, también es necesario argumentar las afirmaciones inversas, a saber, que el número de componentes antarafaciales b + d y el recuento de electrones (4 n + 2 o 4 n) implica la paridad de a + d que viene dada por las reglas de Woodward-Hoffmann (impar para permitido, par para prohibido). Otra ronda de análisis de casos (algo tediosos) mostrará fácilmente que este es el caso.
Para resumir, tenemos la siguiente declaración, que es matemáticamente equivalente a la regla de selección pericíclica generalizada original:
Un proceso pericíclico que involucra 4n+2 o 4n electrones está térmicamente permitido si y solo si el número de componentes antarafaciales involucrados es par o impar, respectivamente.
| huckel | Möbius | |
|---|---|---|
| 4 norte +2 e | Aromático permitido | Antiaromático prohibido |
| 4 no _ | Antiaromático prohibido | Aromático permitido |
En esta formulación, el recuento de electrones se refiere a todo el sistema de reacción, en lugar de a los componentes individuales, como se enumera en la declaración original de Woodward y Hoffmann. En la práctica, un número par o impar de componentes antarafaciales generalmente significa cero o un componente antarafacial, respectivamente, ya que los estados de transición que involucran dos o más componentes antarafaciales generalmente se ven desfavorecidos por la tensión. Como excepciones, ciertas reacciones intramoleculares pueden estar restringidas geométricamente de tal manera que impongan una trayectoria antarafacial para múltiples componentes. Además, en algunos casos, por ejemplo, el reordenamiento de Cope, se puede considerar que la misma geometría de estado de transición (no necesariamente tensa) contiene dos componentes supra o dos antara π, dependiendo de cómo se dibujen las conexiones entre los lóbulos orbitales.
Esta formulación alternativa aclara la equivalencia de las reglas de Woodward-Hoffmann con el análisis de Dewar-Zimmerman (ver más abajo). Un número total par de inversiones de fase equivale a un número par de componentes antarafaciales y corresponde a la topología de Hückel, que requiere 4 n + 2 electrones para la aromaticidad, mientras que un número total impar de inversiones de fase equivale a un número impar de componentes antarafaciales y corresponde a la topología de Möbius, que requiere 4 n electrones para la aromaticidad. Para resumir la teoría del estado de transición aromático: las reacciones pericíclicas térmicas proceden a través de (4 n + 2) -electrones Hückel o (4 n) -electrones Möbius estados de transición.
Como mnemotécnico, la formulación anterior se puede reformular de la siguiente manera:
Un proceso pericíclico de estado fundamental que involucra N pares de electrones y A componentes antarafaciales está permitido por simetría si y solo si N + A es impar.
Prueba alternativa de equivalencia
La equivalencia de las dos formulaciones también puede verse mediante un simple argumento de paridad sin recurrir al análisis de casos.
Proposición. Las siguientes formulaciones de las reglas de Woodward-Hoffmann son equivalentes:
(A) Para una reacción pericíclica, si la suma del número de componentes suprafaciales 4q + 2 y componentes antarafaciales 4r es impar, entonces está térmicamente permitida; de lo contrario, la reacción está térmicamente prohibida.
(B) Para una reacción pericíclica, si el número total de componentes antarafaciales de una reacción de (4n + 2) electrones es par o el número total de componentes antarafaciales de una reacción de 4n electrones es impar, entonces se permite térmicamente; de lo contrario, la reacción está térmicamente prohibida.
Prueba de equivalencia: indexe los componentes de unareacción pericíclica de k


Tenemos una reformulación matemáticamente equivalente de (A):
(A') Se permite térmicamente una colección de símbolos si y solo si el número de símbolos con la propiedad es impar.
Dado que el conteo total de electrones es 4 n + 2 o 4 n precisamente cuando 
(B') Una colección de símbolos está térmicamente permitida si y solo si exactamente uno de o es impar.
Basta mostrar que (A') y (B') son equivalentes. Exactamente uno de 







.
Así,
Por poner un ejemplo concreto, a una hipotética reacción con el descriptor [ π 6 s + π 4 a + π 2 a ] se le asignaría el conjunto {(1, 0, 1), (0, 1, 2), (1, 1, 3)} en el esquema anterior. Hay dos componentes, (1, 0, 1) y (0, 1, 2), con la propiedad 
Ejemplos
Esta formulación para una reacción de 2 componentes es equivalente a las reglas de selección para reacciones de cicloadición [ p + q ] que se muestran en la siguiente tabla:
| pag + q | Térmicamente permitido | Permitido fotoquímicamente |
|---|---|---|
| 4 norte | pags + qa o pags + qs _ _ _ _ | p s + q s o p a + q a |
| 4 n + 2 | p s + q s o p a + q a | pags + qa o pags + qs _ _ _ _ |
Si el número total de electrones es 4 n + 2, entonces uno está en la fila inferior de la tabla. La reacción está térmicamente permitida si es suprafacial con respecto a ambos componentes o antarafacial con respecto a ambos componentes. Es decir, el número de componentes antarafaciales es par (es 0 o 2). De manera similar, si el número total de electrones es 4 n, entonces uno está en la fila superior de la tabla. Está permitido térmicamente si es suprafacial respecto a un componente y antarafacial respecto al otro. Por lo tanto, el número total de componentes antarafaciales siempre es impar, ya que siempre es 1.
Las siguientes son algunas clases de reacción de estado fundamental (es decir, térmicas) comunes analizadas a la luz de las reglas generalizadas de Woodward-Hoffmann.
[2+2] Cicloadición
Una cicloadición [2+2] es un proceso de 4 electrones que reúne dos componentes. Por lo tanto, según las reglas generales de WH anteriores, solo se permite si la reacción es antarafacial con respecto a exactamente un componente. Esta es la misma conclusión a la que se llegó con los diagramas de correlación en la sección anterior.
A la derecha se muestra un ejemplo raro pero estereoquímicamente inequívoco de una cicloadición [ π 2 s + π 2 a ]. La tensión y las propiedades estéricas del doble enlace trans permiten este proceso generalmente cinéticamente desfavorable. También se cree que el cis, trans -1,5-ciclooctadieno experimenta dimerización a través de este modo. Los cetenos son una gran clase de reactivos que favorecen la cicloadición [2 + 2] con olefinas. El análisis MO de la cicloadición de cetena se vuelve complicado y ambiguo por la interacción simultánea pero independiente de los orbitales ortogonales de la cetena, pero puede involucrar un [ π 2 s + π2 a ] interacción también.
[4+2] Cicloadición
La reacción síncrona de Diels-Alder de 6π electrones es una cicloadición [ π 4 s + π 2 s ] (es decir, suprafacial con respecto a ambos componentes), como se ejemplifica en la reacción de la derecha.
Por lo tanto, como el número total de componentes antarafaciales es 0, que es par, la reacción tiene simetría permitida. Esta predicción concuerda con el experimento ya que la reacción de Diels-Alder es una reacción pericíclica bastante fácil.
4 n Reacción electrocíclica
Se puede considerar que una reacción de apertura de anillo electrocíclico de 4 n electrones tiene 2 componentes: el sistema π y la ruptura del enlace σ. Con respecto al sistema π, la reacción es suprafacial. Sin embargo, con un mecanismo conrotatorio, como se muestra en la figura anterior, la reacción es antarafacial con respecto al enlace σ. A la inversa, con un mecanismo disrotatorio, es suprafacial con respecto al enlace σ que se rompe.
Según las reglas anteriores, para una reacción pericíclica de 4 n electrones de 2 componentes, debe haber un componente antarafacial. Por lo tanto, la reacción debe proceder a través de un mecanismo conrotatorio. Esto concuerda con el resultado obtenido en los diagramas de correlación anteriores.
4 n + 2 reacción electrocíclica
Una reacción de apertura de anillo electrocíclica de 4 n + 2 también es una reacción pericíclica de 2 componentes que es suprafacial con respecto al sistema π. Por lo tanto, para que se permita la reacción, el número de componentes antarafaciales debe ser 0, es decir, debe ser suprafacial con respecto a la ruptura del enlace σ también. Por lo tanto, se permite un mecanismo disrotatorio por simetría.
[1, j ] -reordenamiento sigmatrópico
Un reordenamiento [1, j ]-sigmatrópico también es una reacción pericíclica de dos componentes: un componente es el sistema π, el otro componente es el grupo migratorio. El caso más simple es un cambio de [1, j ]-hidruro a través de un sistema π donde j es impar. En este caso, como el hidrógeno tiene solo un orbital s esféricamente simétrico, la reacción debe ser suprafacial con respecto al hidrógeno. El número total de electrones involucrados es (j + 1) ya que hay (j − 1)/2 enlace π más el enlace σ involucrado en la reacción. Si j = 4 n − 1 entonces debe ser antarafacial, y si j = 4 n + 1 entonces debe ser suprafacial. Esto concuerda con el experimento de que los cambios de [1,3]-hidruro generalmente no se observan ya que el proceso antarafacial permitido por simetría no es factible, pero los cambios de [1,5]-hidruro son bastante fáciles.
Para un cambio de [1, j ]-alquilo, donde la reacción puede ser antarafacial (es decir, estereoquímica inversa) con respecto al centro de carbono, se aplican las mismas reglas. Si j = 4 n − 1, entonces la reacción está permitida por simetría si es antarafacial con respecto al sistema π o si invierte la estereoquímica en el carbono. Si j = 4 n + 1, entonces la reacción está permitida por simetría si es suprafacial con respecto al sistema π y retiene la estereoquímica en el centro de carbono.
A la derecha está uno de los primeros ejemplos de un cambio [1,3]-sigmatrópico que se descubrió, informado por Berson en 1967. Para permitir la inversión de la configuración, cuando se rompe el enlace σ, el C(H)(D) el resto se retuerce en el estado de transición, con la hibridación del carbono aproximándose a sp, de modo que el orbital p restante sin hibridar mantiene la superposición con los carbonos 1 y 3.
Equivalencia de otros modelos teóricos
Análisis Dewar-Zimmerman
Las reglas generalizadas de Woodward-Hoffmann, dadas por primera vez en 1969, son equivalentes a un enfoque general anterior, el concepto de Möbius-Hückel de Zimmerman, que se estableció por primera vez en 1966 y también se conoce como teoría del estado de transición aromática. Como principio central, la teoría del estado de transición aromático sostiene que las reacciones pericíclicas 'permitidas' proceden a través de estados de transición con carácter aromático, mientras que las reacciones pericíclicas 'prohibidas' encontrarían estados de transición que son de naturaleza antiaromática. En el análisis de Dewar-Zimmerman, uno se ocupa de la topología del estado de transición de la reacción pericíclica. Si el estado de transición implica 4 nelectrones, la topología de Möbius es aromática y la topología de Hückel es antiaromática, mientras que si el estado de transición involucra 4 n + 2 electrones, la topología de Hückel es aromática y la topología de Möbius es antiaromática. La paridad del número de inversiones de fase (descritas en detalle a continuación) en el estado de transición determina su topología. Una topología de Möbius implica un número impar de inversiones de fase, mientras que una topología de Hückel implica un número par de inversiones de fase.
En relación con la terminología de Woodward-Hoffmann, el número de componentes antarafaciales y el número de inversiones de fase siempre tienen la misma paridad. En consecuencia, un número impar de componentes antarafaciales da la topología de Möbius, mientras que un número par da la topología de Hückel. Por lo tanto, para reafirmar los resultados de la teoría del estado de transición aromático en el lenguaje de Woodward y Hoffmann, una reacción de 4 n - electrones es térmicamente permitida si y solo si tiene un número impar de componentes antarafaciales (es decir, topología de Möbius); una reacción de (4 n + 2) electrones es térmicamente permitida si y solo si tiene un número par de componentes antarafaciales (es decir, topología de Hückel).
Procedimiento para el análisis de Dewar-Zimmerman (ejemplos que se muestran a la derecha): Paso 1. Sombree todos los orbitales básicos que forman parte del sistema pericíclico. El sombreado puede ser arbitrario. En particular, no es necesario que el sombreado refleje la fase de los OM de polieno; cada orbital base simplemente necesita tener dos lóbulos en fases opuestas en el caso de p o sporbitales híbridos, o una sola fase en el caso de un orbital s. Paso 2. Dibuje conexiones entre los lóbulos de los orbitales base que estén bien dispuestos geométricamente para interactuar en el estado de transición. Las conexiones a realizar dependen de la topología del estado de transición. (Por ejemplo, en la figura se muestran diferentes conexiones en los casos de electrociclación con y disrotatoria.) Paso 3. Cuente el número de conexiones que ocurren entre lóbulos de sombreado opuesto: cada una de estas conexiones constituye una inversión de fase. Si el número de inversiones de fase es par, el estado de transición es Hückel, mientras que si el número de inversiones de fase es impar, el estado de transición es Möbius. Paso 4. Concluya que la reacción pericíclica está permitida si el conteo de electrones es 4 n + 2 y el estado de transición es Hückel, o si el conteo de electrones es 4 n y el estado de transición es Möbius; de lo contrario, concluya que la reacción pericíclica está prohibida.
Es importante destacar que cualquier esquema de asignación de fases relativas a los orbitales base es aceptable, ya que invertir la fase de cualquier orbital simple agrega 0 o ±2 inversiones de fase al total, un número par, de modo que la paridad del número de inversiones (número de inversiones módulo 2) no cambia.
Reinterpretación con teoría funcional de la densidad conceptual
Recientemente, las reglas de Woodward-Hoffmann se han reinterpretado utilizando la teoría funcional de densidad conceptual (DFT). La clave del análisis es la función dual descriptor, propuesta por Christophe Morell, André Grand y Alejandro Toro-Labbé, la segunda derivada de la densidad electrónica con respecto al número de electrones. Esta función de respuesta es importante ya que la reacción de dos componentes A y B que implican una transferencia de electrones dependerá de la capacidad de respuesta de la densidad de electrones a la donación o aceptación de electrones, es decir, la derivada de la función de Fukui. De hecho, desde un punto de vista simplista, la función de descriptor dual da una lectura de la electrofilia o nucleofilia de las diversas regiones de la molécula. Para 






Esto tiene un sentido intuitivo, ya que si una región es mejor para aceptar electrones que para donarlos, entonces el LUMO debe dominar y la función de descriptor dual será positiva. Por el contrario, si una región es mejor donando electrones, el término HOMO dominará y el descriptor será negativo. Observe que aunque el concepto de fase y orbitales se reemplaza simplemente por la noción de densidad electrónica, esta función aún toma valores tanto positivos como negativos.
Las reglas de Woodward-Hoffmann se reinterpretan usando esta formulación emparejando interacciones favorables entre regiones de densidad electrónica para las cuales el descriptor dual tiene signos opuestos. Esto es equivalente a maximizar las interacciones favorables predichas y minimizar las interacciones repulsivas. Para el caso de una cicloadición [4+2], se muestra un esquema simplificado de los reactivos con la función de descriptor dual coloreada (rojo=positivo, azul=negativo) en la configuración supra/supra óptima a la izquierda. Este método predice correctamente las reglas WH para las principales clases de reacciones pericíclicas.
Excepciones
En el Capítulo 12 de The Conservation of Orbital Symmetry, titulado "Violaciones", Woodward y Hoffmann declararon:
¡No hay ninguno! Tampoco cabe esperar violaciones de un principio tan fundamental de máxima vinculación.
A pesar de este pronunciamiento, es importante reconocer que las reglas de Woodward-Hoffmann se utilizan para predecir las alturas relativas de las barreras y, por lo tanto, los posibles mecanismos de reacción, y que solo tienen en cuenta las barreras debido a la conservación de la simetría orbital. Por lo tanto, no se garantiza que una reacción permitida por la simetría WH realmente tenga lugar de una manera fácil. Por el contrario, es posible, con suficiente aporte energético, lograr un producto anti-Woodward-Hoffmann. Esto es especialmente frecuente en sistemas estéricamente restringidos, donde el producto WH tiene una barrera estérica adicional que superar. Por ejemplo, en la apertura electrocíclica del anillo del derivado dimetilbiciclo[0.2.3]hepteno (1), no es posible un mecanismo conrotatorio debido a la tensión angular resultante y la reacción avanza lentamente a través de un mecanismo disrotatorio a 400 C para dar un producto de cicloheptadieno. También se pueden observar violaciones en casos con fuerzas impulsoras termodinámicas muy fuertes. La descomposición de dioxetano-1,2-diona en dos moléculas de dióxido de carbono, famosa por su papel en la luminiscencia de las barras luminosas, se ha analizado computacionalmente. En ausencia de fluorescentes, ahora se cree que la reacción procede de manera concertada (aunque asíncrona), a través de una retro-[2+2]-cicloadición que viola formalmente las reglas de Woodward-Hoffmann.
De manera similar, un artículo reciente describe cómo se puede usar el estrés mecánico para remodelar las vías de reacción química para generar productos que aparentemente violan las reglas de Woodward-Hoffman. En este artículo, utilizan la irradiación de ultrasonido para inducir una tensión mecánica en los polímeros funcionalizados con enlaces unidos sin o anti en el anillo de ciclobuteno. Los estudios computacionales predicen que la fuerza mecánica, resultante de la fricción de los polímeros, induce el alargamiento del enlace a lo largo de la coordenada de reacción del mecanismo conrotatorio en el ciclobuteno anti-bisustituido, y a lo largo de la coordenada de reacción del mecanismo disrotatorio en el ciclobuteno bisustituido sin.. Así, en el ciclobuteno bisustituido sin, se predice que se formará el producto anti -WH.
Esta predicción computacional fue respaldada por experimentos en el sistema a continuación. Los polímeros funcionalizados con enlace se conjugaron con benzociclobuteno cis en conformaciones sin y anti. Como se había predicho, ambos productos dieron el mismo producto (Z,Z) según lo determinado por inactivación mediante una reacción estereoespecífica de Diels-Alder con la maleimida sustituida. En particular, el producto sustituido con syn dio el producto anti-WH, presumiblemente porque el estiramiento mecánico a lo largo de la coordenada de la vía disrotatoria redujo la barrera de la reacción bajo la vía disrotatoria lo suficiente como para sesgar ese mecanismo.

Controversia
Se ha afirmado que Elias James Corey, también ganador del Premio Nobel, se siente responsable de las ideas que sentaron las bases de esta investigación, y que Woodward se olvidó injustamente de darle crédito por el descubrimiento. En una memoria de 2004 publicada en el Journal of Organic Chemistry, Corey afirma que la idea es prioritaria: "El 4 de mayo de 1964, le sugerí a mi colega RB Woodward una explicación simple que involucraba la simetría de los orbitales moleculares perturbados (HOMO). para las conversiones estereoselectivas de ciclobuteno a 1,3-butadieno y de 1,3,5-hexatrieno a ciclohexadieno que proporcionaron la base para el desarrollo posterior de estas ideas en lo que se conoció como las reglas de Woodward-Hoffmann".
Corey, entonces de 35 años, estaba trabajando hasta la noche del lunes 4 de mayo, como solían hacer él y los otros químicos motivados. Alrededor de las 8:30 pm, pasó por la oficina de Woodward, y Woodward planteó una pregunta sobre cómo predecir el tipo de anillo que formaría una cadena de átomos. Después de alguna discusión, Corey propuso que la configuración de los electrones gobernaba el curso de la reacción. Woodward insistió en que la solución no funcionaría, pero Corey dejó dibujos en la oficina, seguro de que estaba en lo cierto.
"Sentí que esto iba a ser un desarrollo realmente interesante y esperaba algún tipo de empresa conjunta", escribió. Pero al día siguiente, Woodward voló a la oficina de Corey cuando él y un colega se iban a almorzar y presentó la idea de Corey como propia, y luego se fue. Corey estaba atónito.
En una refutación de 2004 publicada en Angewandte Chemie, Roald Hoffmann negó la afirmación: cita a Woodward de una conferencia pronunciada en 1966 diciendo:"RECUERDO muy claramente, y todavía me sorprende un poco, que el destello crucial de iluminación me llegó en forma algebraica, en lugar de pictórica o geométrica. De la nada, se me ocurrió que los coeficientes de los términos terminales en la expresión matemática que representaba el orbital molecular ocupado más alto del butadieno eran de signo opuesto, mientras que las de la expresión correspondiente para el hexatrieno poseían el mismo signo. Desde aquí no había más que un pequeño paso hasta la visión geométrica, y más obviamente químicamente relevante, de que en En la ciclación interna de un dieno, la cara superior de un átomo terminal debería atacar la cara inferior del otro, mientras que en el caso del trieno, la formación de un nuevo enlace debería involucrar a las caras superiores (o pari passu, la inferior) de ambos. átomos terminales".
Además, Hoffmann señala que en dos publicaciones de 1963 y 1965, Corey describió una síntesis total del compuesto dihidrocostunólido. Aunque describen una reacción electrocíclica, Corey no tiene nada que ofrecer con respecto a explicar la estereoespecificidad de la síntesis.
Esta reacción fotoquímica que involucra 6 = 4 × 1 + 2 electrones ahora se reconoce como conrotatoria.
Contenido relacionado
Teoría de Marcus
Louis Pasteur
Fenol