
Teoría de Marcus
La teoría de Marcus es una teoría desarrollada originalmente por Rudolph A. Marcus, a partir de 1956, para explicar las velocidades de las reacciones de transferencia de electrones: la velocidad a la que un electrón puede moverse o saltar de una especie química (llamada donante de electrones) a otra (llamada el aceptor de electrones). Originalmente se formuló para abordar las reacciones de transferencia de electrones de la esfera exterior, en las que las dos especies químicas solo cambian de carga con un salto de electrones (p. ej., la oxidación de un ion como Fe /Fe), pero no experimentan grandes cambios estructurales. Se amplió para incluir las contribuciones de transferencia de electrones de la esfera interna, en las que se tiene en cuenta un cambio de distancias o geometría en las capas de solvatación o coordinación de las dos especies químicas (las distancias Fe-O en Fe(H 2O) y Fe(H 2 O) son diferentes).
Para las reacciones de transferencia de electrones sin formar o romper enlaces, la teoría de Marcus reemplaza a la teoría del estado de transición de Eyring, que se ha derivado para reacciones con cambios estructurales. Ambas teorías conducen a ecuaciones de tasa de la misma forma exponencial. Sin embargo, mientras que en la teoría de Eyring los compañeros de reacción se acoplan fuertemente en el curso de la reacción para formar un complejo activado estructuralmente definido, en la teoría de Marcus están débilmente acoplados y conservan su individualidad. Es la reorganización inducida térmicamente de los alrededores, el solvente (esfera exterior) y la envoltura del solvente o los ligandos (esfera interior) lo que crea la situación geométricamente favorable previa e independiente del salto de electrones.
La teoría clásica original de Marcus para las reacciones de transferencia de electrones en la esfera externa demuestra la importancia del solvente y conduce al cálculo de la energía de activación libre de Gibbs, usando las propiedades de polarización del solvente, el tamaño de los reactivos, la distancia de transferencia y la energía libre de Gibbs 
RA Marcus recibió el Premio Nobel de Química en 1992 por esta teoría. La teoría de Marcus se utiliza para describir una serie de procesos importantes en química y biología, incluida la fotosíntesis, la corrosión, ciertos tipos de quimioluminiscencia, la separación de carga en algunos tipos de células solares y más. Además de las aplicaciones de la esfera interior y exterior, la teoría de Marcus se ha ampliado para abordar la transferencia de electrones heterogéneos.
ET exterior vs interior
En una reacción redox, un donante de electrones D debe difundirse hacia el aceptor A, formando un complejo precursor, que es lábil pero permite la transferencia de electrones para dar un complejo sucesor. Luego, la pareja se disocia. Para una transferencia de un electrón, la reacción es
![{displaystyle {ce {{D}+A<=>[k_{12}][k_{21}][D{dotsm }A]<=>[k_{23}][k_{32}] [D+{dotsm }A^{-}]->[k_{30}]{D+}+{A^{-}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9011d1fe6f8639a87bf5c31e70967d0fcdf4bcc1)
(Es posible que D y A ya tengan cargos). Aquí k 12, k 21 y k 30 son constantes de difusión, k 23 y k 32constantes de velocidad de las reacciones activadas. La reacción total puede estar controlada por difusión (el paso de transferencia de electrones es más rápido que la difusión, cada encuentro conduce a una reacción) o controlada por activación (se alcanza el "equilibrio de asociación", el paso de transferencia de electrones es lento, la separación del complejo sucesor es rápido). Las capas de ligando alrededor de A y D se conservan. Este proceso se llama transferencia de electrones en la esfera exterior. La esfera exterior ET es el foco principal de la teoría tradicional de Marcus. El otro tipo de reacciones redox es la esfera interna donde A y D están unidos covalentemente por un ligando puente. Las tasas de tales reacciones ET dependen de las tasas de intercambio de ligandos.
El problema
En las reacciones redox de la esfera exterior no se forman ni se rompen enlaces; sólo tiene lugar una transferencia de electrones (ET). Un ejemplo bastante simple es la reacción redox Fe /Fe, la reacción de autointercambio que se sabe que siempre ocurre en una solución acuosa que contiene tanto FeSO 4 como Fe 2 (SO 4) 3 (por supuesto, con tasas iguales y medibles en ambos direcciones y con la energía de reacción libre de Gibbs 
A partir de la dependencia de la temperatura de la velocidad de reacción se determina una energía de activación, y esta energía de activación se interpreta como la energía del estado de transición en un diagrama de reacción. Este último se dibuja, según Arrhenius y Eyring, como un diagrama de energía con la coordenada de reacción como abscisa. La coordenada de reacción describe la ruta de energía mínima desde los reactivos hasta los productos, y los puntos de esta coordenada son combinaciones de distancias y ángulos entre y en los reactivos en el curso de la formación y/o ruptura de enlaces. El máximo del diagrama de energía, el estado de transición, se caracteriza por una configuración específica de los átomos. Además, en el TST de Eyringun cambio bastante específico de las coordenadas nucleares es responsable de cruzar el punto máximo, por lo que una vibración en esta dirección se trata como una traslación.
Para las reacciones redox de la esfera exterior no puede haber tal camino de reacción, pero sin embargo se observa una energía de activación. La ecuación de velocidad para las reacciones controladas por activación tiene la misma forma exponencial que la ecuación de Eyring,


El modelo marcus
La consecuencia de una transferencia de electrones es el reordenamiento de las cargas, y esto influye en gran medida en el entorno del solvente. Porque las moléculas dipolares del solvente se reorganizan en la dirección del campo de las cargas (esto se llama polarización de orientación), y también los átomos y electrones en las moléculas del solvente están ligeramente desplazados (polarización atómica y electrónica, respectivamente). Es esta polarización del disolvente la que determina la energía libre de activación y, por tanto, la velocidad de reacción.
Las reacciones de sustitución, eliminación e isomerización difieren de la reacción redox de la esfera externa no solo en los cambios estructurales descritos anteriormente, sino también en el hecho de que tienen lugar los movimientos de los núcleos y el cambio de cargas (transferencia de carga, CT) en el camino de las reacciones. de forma continua y concertada: las configuraciones nucleares y la distribución de carga están siempre "en equilibrio". Esto se ilustra con la sustitución S N 2 de la saponificación de un haluro de alquilo donde el ataque del lado posterior del ion OH expulsa un ion haluro y donde se debe visualizar un estado de transición con un átomo de carbono de cinco coordenadas. El sistema de reactivos se acopla tan estrechamente durante la reacción que forman el complejo activado como una entidad integral. El solvente aquí tiene un efecto menor.
Por el contrario, en las reacciones redox de la esfera externa, el desplazamiento de los núcleos en los reactivos es pequeño, aquí el solvente tiene el papel dominante. El acoplamiento donador-aceptor es débil, ambos mantienen su identidad durante la reacción. Por lo tanto, el electrón, al ser una partícula elemental, solo puede "saltar" como un todo (transferencia de electrones, ET). Si el electrón salta, la transferencia es mucho más rápida que el movimiento de las grandes moléculas del solvente, con la consecuencia de que las posiciones nucleares de los compañeros de reacción y las moléculas del solvente son las mismas antes y después del salto del electrón (principio de Franck-Condon). El salto del electrón se rige por las reglas de la mecánica cuántica, solo es posible si además la energía del sistema ET no cambia "durante" el salto.
La disposición de las moléculas de disolvente depende de la distribución de carga de los reactivos. Si la configuración del solvente debe ser la misma antes y después del salto y la energía no puede cambiar, entonces el solvente no puede estar en el estado de solvatación del precursor ni en el del complejo sucesor ya que son diferentes, tiene que estar en algún lugar en Entre. Para la reacción de autointercambio por razones de simetría, una disposición de las moléculas de disolvente exactamente en el medio de las del complejo precursor y sucesor cumpliría las condiciones. Esto significa que la disposición del solvente con la mitad del electrón tanto en el donante como en el aceptor sería el entorno correcto para el salto. Además, en este estado la energía del precursor y del sucesor en su entorno solvente sería la misma.
Sin embargo, el electrón como partícula elemental no se puede dividir, reside en el donante o en el aceptor y dispone las moléculas del disolvente en equilibrio. El "estado de transición", por otro lado, requiere una configuración solvente que resultaría de la transferencia de medio electrón, lo cual es imposible. Esto significa que la distribución de carga real y la polarización del solvente requerida no están en un "equilibrio". Sin embargo, es posible que el solvente tome una configuración correspondiente al "estado de transición", incluso si el electrón se encuentra en el donante o el aceptor. Esto, sin embargo, requiere energía. Esta energía puede ser proporcionada por la energía térmica del solvente y las fluctuaciones térmicas pueden producir el estado de polarización correcto. Una vez que se ha alcanzado esto, el electrón puede saltar.de la disposición correcta del solvente y el salto de electrones están desacoplados y no ocurren en un proceso sincrónico. Por lo tanto, la energía del estado de transición es principalmente energía de polarización del solvente.
Teoría de marcus
El sistema macroscópico: dos esferas conductoras
Sobre la base de su razonamiento, RA Marcus desarrolló una teoría clásica con el objetivo de calcular la energía de polarización de dicho estado de no equilibrio. De la termodinámica es bien sabido que la energía de tal estado se puede determinar si se encuentra un camino reversible a ese estado. Marcus logró encontrar ese camino a través de dos pasos de carga reversibles para la preparación del "estado de transición" del complejo precursor.
Cuatro elementos son esenciales para el modelo en el que se basa la teoría:
- Marcus emplea un modelo clásico, puramente electrostático. La carga (muchas cargas elementales) puede transferirse en cualquier parte de un cuerpo a otro.
- Marcus separa la polarización rápida de electrones P e y la polarización lenta de átomos y orientación P u del solvente sobre la base de que sus constantes de tiempo difieren en varios órdenes de magnitud.
- Marcus separa la esfera interior (reactivo + moléculas de disolvente fuertemente unidas, en complejos + ligandos) y la esfera exterior (disolvente libre)
- En este modelo, Marcus se limita a calcular la energía de la esfera exterior de la polarización de no equilibrio del "estado de transición". La energía de la esfera exterior suele ser mucho mayor que la contribución de la esfera interior debido a las fuerzas electrostáticas de largo alcance (compárese con la teoría electroquímica de Debye-Hückel).
La herramienta de Marcus es la teoría de la polarización dieléctrica en solventes. Resolvió el problema de forma general para una transferencia de carga entre dos cuerpos de forma arbitraria con superficie y volumen de carga arbitrarios. Para la reacción de autointercambio, el par redox (por ejemplo, Fe(H 2 O) 6 / Fe(H 2 O) 6) se sustituye por dos esferas conductoras macroscópicas a una distancia definida que transportan cargas específicas. Entre estas esferas se intercambia reversiblemente una cierta cantidad de carga.
En el primer paso, se calcula la energía W I de la transferencia de una cantidad específica de carga, por ejemplo, para el sistema en un estado en el que ambas esferas llevan la mitad de la cantidad de carga que debe transferirse. Este estado del sistema se puede alcanzar transfiriendo la carga respectiva de la esfera donante al vacío y luego de vuelta a la esfera aceptora. Luego, las esferas en este estado de carga dan lugar a un campo eléctrico definido en el solvente que crea la polarización total del solvente P u + P e. Del mismo modo, esta polarización del solvente interactúa con las cargas.
En un segundo paso, se calcula la energía W II de la transferencia reversible (hacia atrás) de la carga a la primera esfera, nuevamente a través del vacío. Sin embargo, la polarización del átomo y la orientación P u se mantienen fijas, solo la polarización electrónica P e puede ajustarse al campo de la nueva distribución de carga y la P u fija. Después de este segundo paso, el sistema está en el estado deseado con una polarización electrónica correspondiente al punto de partida de la reacción redox y una polarización atómica y de orientación correspondiente al "estado de transición". La energía W I + W II de este estado es, termodinámicamente hablando, una energía libre de Gibbs G.
Por supuesto, en este modelo clásico es posible la transferencia de cualquier cantidad arbitraria de carga Δe. Entonces, la energía del estado de no equilibrio y, en consecuencia, de la energía de polarización del solvente, se puede probar como una función de Δe. Así, Marcus ha agrupado, de una manera muy elegante, las coordenadas de todas las moléculas de solvente en una única coordenada de polarización del solvente Δp que está determinada por la cantidad de carga transferida Δe. Entonces llegó a una simplificación de la representación de la energía a solo dos dimensiones: G = f(Δe). El resultado de dos esferas conductoras en un solvente es la fórmula de Marcus

Donde r 1 y r 2 son los radios de las esferas y R es su separación, ε s y ε opt son las constantes dieléctricas estática y de alta frecuencia (óptica) del solvente, Δe la cantidad de carga transferida. La gráfica de G frente a Δe es una parábola (Fig. 1). En la teoría de Marcus, la energía correspondiente a la transferencia de una unidad de carga (Δe = 1) se denomina energía de reorganización (esfera exterior) λ o, es decir, la energía de un estado en el que la polarización correspondería a la transferencia de una unidad de cantidad de carga., pero la distribución de carga real es la anterior a la transferencia. En términos de dirección de intercambio, el sistema es simétrico.
El sistema microscópico: el par donante-aceptor
Reducir el modelo de dos esferas al nivel molecular crea el problema de que en la reacción de autointercambio la carga ya no puede transferirse en cantidades arbitrarias, sino solo como un solo electrón. Sin embargo, la polarización todavía está determinada por el conjunto total de las moléculas de disolvente y, por lo tanto, todavía puede tratarse de forma clásica, es decir, la energía de polarización no está sujeta a limitaciones cuánticas. Por lo tanto, la energía de reorganización del solvente puede calcularse como debida a un hipotéticotransferencia y transferencia inversa de una carga elemental parcial según la fórmula de Marcus. Así, la energía de reorganización para las reacciones químicas redox, que es una energía libre de Gibbs, es también una función parabólica de Δe de esta transferencia hipotética. Para la reacción de autointercambio, donde por razones de simetría Δe = 0,5, la energía libre de activación de Gibbs es ΔG (0) = λ o /4 (ver Fig. 1 y Fig. 2 intersección de las parábolas I y f, f(0), respectivamente).
Hasta ahora todo era física, ahora entra algo de química. La reacción de autointercambio es una reacción redox muy específica, la mayoría de las reacciones redox son entre diferentes socios, por ejemplo
![{displaystyle {ce {{[Fe^{II}(CN)6]^{4-}}+{[Ir^{IV}Cl6]^{2-}}<=>{[Fe^{III }(CN)6]^{3-}}+{[Ir^{III}Cl6]^{3-}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/31656dcef696dc00f999db7bf1e70e8747989c22)
y tienen energías de reacción libres de Gibbs positivas (endergónicas) o negativas (exergónicas)
Como los cálculos de Marcus se refieren exclusivamente a las propiedades electrostáticas en el solvente (esfera exterior) 


Cálculos simples de los puntos de intersección de las parábolas i, 






La intersección de esas parábolas representa una energía de activación y no la energía de un estado de transición de configuración fija de todos los núcleos del sistema como es el caso de la sustitución y otras reacciones mencionadas. El estado de transición de las últimas reacciones tiene que cumplir con las condiciones estructurales y energéticas, las reacciones redox solo tienen que cumplir con el requisito de energía. Mientras que la geometría del estado de transición en las demás reacciones es la misma para todos los pares de reactivos, para los pares redox muchos entornos de polarización pueden cumplir las condiciones energéticas.
La fórmula de Marcus muestra una dependencia cuadrática de la energía libre de activación de Gibbs con respecto a la energía libre de reacción de Gibbs. Es un conocimiento general de la gran cantidad de experiencia química que las reacciones suelen ser más rápidas cuanto más negativas son 
El máximo de la tasa de ET se espera en 


Transferencia de electrones en la esfera interna
En el modelo de la esfera exterior, se consideró que el donante o el aceptor y las capas de solvatación fuertemente unidas o los ligandos complejos formaban estructuras rígidas que no cambian en el curso de la transferencia de electrones. Sin embargo, las distancias en la esfera interna dependen de la carga del donante y del aceptor, por ejemplo, las distancias entre el ión central y el ligando son diferentes en los complejos que llevan diferentes cargas y nuevamente se debe obedecer el principio de Franck-Condon: para que ocurra el salto del electrón., los núcleos tienen que tener una configuración idéntica tanto al complejo precursor como al sucesor, por supuesto muy distorsionada. En este caso, el requisito de energía se cumple automáticamente.
En este caso de esfera interior, se mantiene el concepto de Arrhenius, el estado de transición de estructura geométrica definida se alcanza a lo largo de una coordenada de reacción geométrica determinada por movimientos nucleares. No se necesita más movimiento nuclear para formar el complejo sucesor, solo los saltos de electrones, lo que hace una diferencia en la teoría TST. La coordenada de reacción para la energía de la esfera interna está gobernada por vibraciones y difieren en las especies oxidadas y reducidas.
Para el sistema de autointercambio Fe /Fe solo se considera la vibración de respiración simétrica de las seis moléculas de agua alrededor de los iones de hierro. Asumiendo condiciones armónicas esta vibración tiene frecuencias 



donde q 0 es la coordenada normal de equilibrio y 
Las coordenadas normales de equilibrio difieren en Fe(H 2 O) 6 y Fe(H 2 O) 6. Mediante la excitación térmica de la vibración respiratoria se puede alcanzar una geometría que es común tanto para el donante como para el receptor, es decir, las curvas de energía potencial de las vibraciones respiratorias de D y A se cruzan aquí. Esta es la situación en la que el electrón puede saltar. La energía de este estado de transición es la energía de reorganización de la esfera interna λ en.
Para la reacción de autointercambio se puede calcular la distancia metal-agua en el estado de transición

Esto le da a la energía de reorganización de la esfera interna.

Es una suerte que las expresiones de las energías para la reorganización externa e interna tengan la misma forma cuadrática. Las energías de reorganización de la esfera interior y la esfera exterior son independientes, por lo que se pueden sumar para dar 

Aquí, se puede ver que A representa la probabilidad de salto de electrones, exp[-Δ G in / kT ] la de alcanzar el estado de transición de la esfera interna y exp[-Δ G o / kT ] la de ajuste de la esfera externa.
Para reacciones asimétricas (cruzadas) como
![{displaystyle {ce {{[Fe(H2O)6]^{2}+}+{[Co(H2O)6]^{3}+}<=>{[Fe(H2O)6]^{3 }+}+{[Co(H2O)6]^{2}+}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6cc3b6ef06cdd44100c25f3909ffaf574da148c0)
la expresión para 
La probabilidad del salto de electrones.
La fuerza del acoplamiento electrónico del donante y el aceptor decide si la reacción de transferencia de electrones es adiabática o no adiabática. En el caso no adiabático, el acoplamiento es débil, es decir, H AB en la Fig. 3 es pequeño en comparación con la energía de reorganización y el donante y el aceptor conservan su identidad. El sistema tiene cierta probabilidad de saltar de la curva de energía potencial inicial a la final. En el caso adiabático el acoplamiento es considerable, la brecha de 2 H AB es mayor y el sistema permanece en la curva de energía potencial más baja.
La teoría de Marcus, como se expuso anteriormente, representa el caso no adiabático. En consecuencia, se puede aplicar la teoría semiclásica de Landau-Zener, que da la probabilidad de interconversión de donante y aceptor para un solo paso del sistema por la región de intersección de las curvas de energía potencial
![{displaystyle P_{if}=1-exp left[-{frac {4pi ^{2}{H_{if}^{2}}}{hvleft|s_{i}-s_{ f}derecho|}}derecho]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/859b6031bec85625808c0da2d23506517c8fb52f)
donde H if es la energía de interacción en la intersección, v la velocidad del sistema a través de la región de intersección, s i y s f las pendientes allí.
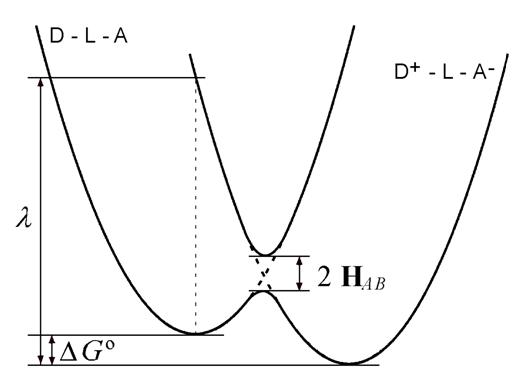
Fig. 3 Diagrama de energía para la transferencia de electrones que incluye la reorganización de las esferas interna y externa y el acoplamiento electrónico: el eje vertical es la energía libre y el eje horizontal es la "coordenada de reacción", un eje simplificado que representa el movimiento de todos los núcleos atómicos (incluido reorganización solvente)
Resolviendo esto, se llega a la ecuación básica de la teoría de Marcus

donde 




Por lo tanto, la teoría de Marcus se basa en la ecuación tradicional de Arrhenius para las velocidades de las reacciones químicas de dos maneras: 1. Proporciona una fórmula para la energía de activación, basada en un parámetro llamado energía de reorganización, así como la energía libre de Gibbs. La energía de reorganización se define como la energía requerida para "reorganizar" la estructura del sistema desde las coordenadas iniciales hasta las finales, sin realizar la transferencia de carga. 2. Proporciona una fórmula para el factor preexponencial en la ecuación de Arrhenius, basada en el acoplamiento electrónico entre el estado inicial y final de la reacción de transferencia de electrones (es decir, la superposición de las funciones de onda electrónicas de los dos estados).
Resultados experimentales
Marcus publicó su teoría en 1956. Durante muchos años hubo una intensa búsqueda de la región invertida que sería una prueba de la teoría. Pero todos los experimentos con series de reacciones de ΔG cada vez más negativos revelaron solo un aumento de la velocidad de reacción hasta el límite de difusión, es decir, hasta un valor que indica que cada encuentro conduce a la transferencia de electrones, y ese límite se mantiene también para valores de ΔG muy negativos. (Comportamiento de Rehm-Weller). Pasaron unos 30 años hasta que Miller, Calcaterra y Closs demostraron inequívocamente que la región invertida era una transferencia de electrones intramolecular en una molécula en la que el donante y el aceptor se mantienen a una distancia constante por medio de un espaciador rígido (Fig. 4).
A posteriori se puede suponer que en los sistemas donde los compañeros de reacción pueden difundirse libremente se puede buscar la distancia óptima para el salto de electrones, es decir, la distancia para la cual ΔG = 0 y ΔG = - λ o. Para λ o depende de R, λ o aumenta para R más grande y la apertura de la parábola más pequeña. Formalmente, siempre es posible cerrar la parábola de la Fig. 2 hasta tal punto que la parábola f interseque la parábola i en el vértice. Entonces siempre ΔG = 0 y la tasa k alcanza el valor difusional máximo para todo ΔG muy negativo. Hay, sin embargo, otros conceptos para el fenómeno,por ejemplo, la participación de estados excitados o que la disminución de las constantes de velocidad estaría tan lejos en la región invertida que escapa a la medición.
RA Marcus y sus colaboradores han desarrollado aún más la teoría descrita aquí en varios aspectos. Han incluido, entre otros, aspectos estadísticos y efectos cuánticos, han aplicado la teoría a la quimioluminiscencia y reacciones de electrodos. RA Marcus recibió el Premio Nobel de Química en 1992, y su Conferencia Nobel ofrece una visión amplia de su trabajo.
Contenido relacionado
Magnesio
Ácido aspártico
Éster