
Sustitución nucleófila acílica
La sustitución nucleófila de acilo describe una clase de reacciones de sustitución que involucran nucleófilos y compuestos de acilo. En este tipo de reacción, un nucleófilo, como un alcohol, una amina o un enolato, desplaza el grupo saliente de un derivado de acilo, como un haluro, anhídrido o éster de ácido. El producto resultante es un compuesto que contiene carbonilo en el que el nucleófilo ha ocupado el lugar del grupo saliente presente en el derivado acilo original. Debido a que los derivados de acilo reaccionan con una amplia variedad de nucleófilos, y debido a que el producto puede depender del tipo particular de derivado de acilo y nucleófilo involucrado, las reacciones de sustitución de acilo nucleófilo pueden usarse para sintetizar una variedad de productos diferentes.
Mecanismo de reacción
Los compuestos de carbonilo reaccionan con los nucleófilos a través de un mecanismo de adición: el nucleófilo ataca al carbono del carbonilo, formando un intermedio tetraédrico. Esta reacción puede ser acelerada por condiciones ácidas, que hacen que el carbonilo sea más electrofílico, o condiciones básicas, que proporcionan un nucleófilo más aniónico y, por lo tanto, más reactivo. El intermedio tetraédrico en sí puede ser un alcohol o un alcóxido, dependiendo del pH de la reacción.
El intermedio tetraédrico de un compuesto de acilo contiene un sustituyente unido al carbono central que puede actuar como grupo saliente. Después de que se forma el intermedio tetraédrico, colapsa, recreando el enlace carbonilo C=O y expulsando el grupo saliente en una reacción de eliminación. Como resultado de este proceso de adición/eliminación de dos pasos, el nucleófilo toma el lugar del grupo saliente en el compuesto carbonilo por medio de un estado intermedio que no contiene un carbonilo. Ambos pasos son reversibles y, como resultado, las reacciones de sustitución de acilo nucleófilo son procesos de equilibrio. Debido a que el equilibrio favorecerá al producto que contiene el mejor nucleófilo, el grupo saliente debe ser un nucleófilo comparativamente pobre para que la reacción sea práctica.
Condiciones ácidas
En condiciones ácidas, el grupo carbonilo del compuesto acilo 1 se protona, lo que lo activa hacia el ataque nucleofílico. En el segundo paso, el carbonilo protonado 2 es atacado por un nucleófilo (H−Z) para dar el intermedio tetraédrico 3. La transferencia de protones del nucleófilo (Z) al grupo saliente (X) da 4, que luego colapsa para expulsar el grupo saliente protonado (H−X), dando el compuesto carbonilo protonado 5. La pérdida de un protón da el producto de sustitución, 6. Debido a que el último paso implica la pérdida de un protón, las reacciones de sustitución de acilo nucleófilo se consideran catalíticas en ácido. También tenga en cuenta que en condiciones ácidas, un nucleófilo existirá normalmente en su forma protonada (es decir, H−Z en lugar de Z).

Condiciones básicas
En condiciones básicas, un nucleófilo (Nuc) ataca al grupo carbonilo del compuesto de acilo 1 para dar el intermedio alcóxido tetraédrico 2. El intermedio colapsa y expulsa el grupo saliente (X) para dar el producto de sustitución 3. Mientras que las reacciones nucleófilas de sustitución de acilo pueden ser catalizadas por bases, la reacción no ocurrirá si el grupo saliente es una base más fuerte que el nucleófilo (es decir, el grupo saliente debe tener un p Ka más alto que el nucleófilo). A diferencia de los procesos catalizados por ácidos, tanto el nucleófilo como el grupo saliente existen como aniones en condiciones básicas.

Este mecanismo está respaldado por experimentos de etiquetado de isótopos. Cuando el propionato de etilo con un grupo etoxi marcado con oxígeno-18 se trata con hidróxido de sodio (NaOH), la marca de oxígeno-18 está completamente ausente del ácido propiónico y se encuentra exclusivamente en el etanol.

Tendencias de reactividad
Hay cinco tipos principales de derivados de acilo. Los haluros de ácido son los más reactivos con los nucleófilos, seguidos de los anhídridos, los ésteres y las amidas. Los iones carboxilato son esencialmente no reactivos frente a la sustitución nucleófila, ya que no poseen grupo saliente. La reactividad de estas cinco clases de compuestos cubre un amplio rango; las velocidades de reacción relativas de los cloruros de ácido y las amidas difieren en un factor de 10.

Un factor importante para determinar la reactividad de los derivados de acilo es la capacidad de grupo saliente, que está relacionada con la acidez. Las bases débiles son mejores grupos salientes que las bases fuertes; una especie con un ácido conjugado fuerte (p. ej., ácido clorhídrico) será un mejor grupo saliente que una especie con un ácido conjugado débil (p. ej., ácido acético). Por lo tanto, el ion cloruro es un mejor grupo saliente que el ion acetato. La reactividad de los compuestos de acilo hacia los nucleófilos disminuye a medida que aumenta la basicidad del grupo saliente, como muestra la tabla.
| Nombre compuesto | Estructura | Dejando el grupo | p K a del ácido conjugado |
|---|---|---|---|
| Cloruro de acetilo |  | −7 | |
| Anhídrido acético | 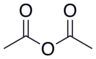 |  | 4.76 |
| Acetato de etilo |  | 15.9 | |
| Acetamida |  | 38 | |
| anión acetato | | N / A | N / A |
Otro factor que juega un papel en la determinación de la reactividad de los compuestos de acilo es la resonancia. Las amidas exhiben dos formas principales de resonancia. Ambos son los principales contribuyentes a la estructura general, tanto que el enlace amida entre el carbono del carbonilo y el nitrógeno de la amida tiene un carácter de doble enlace significativo. La barrera de energía para la rotación alrededor de un enlace amida es de 75 a 85 kJ/mol (18 a 20 kcal/mol), mucho mayor que los valores observados para los enlaces simples normales. Por ejemplo, el enlace C-C en el etano tiene una barrera energética de solo 12 kJ/mol (3 kcal/mol). Una vez que ataca un nucleófilo y se forma un intermedio tetraédrico, se pierde el efecto de resonancia energéticamente favorable. Esto ayuda a explicar por qué las amidas son uno de los derivados de acilo menos reactivos.
Los ésteres exhiben menos estabilización de resonancia que las amidas, por lo que la formación de un intermedio tetraédrico y la subsiguiente pérdida de resonancia no es tan energéticamente desfavorable. Los anhídridos experimentan una estabilización de resonancia aún más débil, ya que la resonancia se divide entre dos grupos carbonilo y son más reactivos que los ésteres y las amidas. En los haluros de ácido, hay muy poca resonancia, por lo que la penalización energética por formar un intermedio tetraédrico es pequeña. Esto ayuda a explicar por qué los haluros de ácido son los derivados de acilo más reactivos.
Reacciones de derivados de acilo
Muchas reacciones nucleófilas de sustitución de acilo implican la conversión de un derivado de acilo en otro. En general, las conversiones entre derivados de acilo deben pasar de un compuesto relativamente reactivo a uno menos reactivo para que sean prácticas; un cloruro de ácido se puede convertir fácilmente en un éster, pero convertir un éster directamente en un cloruro de ácido es esencialmente imposible. Al convertir entre derivados de acilo, el producto siempre será más estable que el compuesto de partida.
También son posibles las reacciones de sustitución nucleófila de acilo que no implican la interconversión entre derivados de acilo. Por ejemplo, las amidas y los ácidos carboxílicos reaccionan con los reactivos de Grignard para producir cetonas. Aquí se presenta una descripción general de las reacciones en las que puede participar cada tipo de derivado de acilo.
Haluros de ácido
Los haluros de ácido son los derivados de acilo más reactivos y se pueden convertir fácilmente en cualquiera de los otros. Los haluros de ácido reaccionarán con los ácidos carboxílicos para formar anhídridos. Si la estructura del ácido y del cloruro de ácido es diferente, el producto es un anhídrido mixto. Primero, el ácido carboxílico ataca al cloruro de ácido (1) para dar el intermedio tetraédrico 2. El intermedio tetraédrico colapsa, expulsando iones de cloruro como grupo saliente y formando especies de oxonio 3. La desprotonación da el anhídrido mixto, 4, y un equivalente de HCl.

Los alcoholes y las aminas reaccionan con haluros de ácido para producir ésteres y amidas, respectivamente, en una reacción conocida formalmente como reacción de Schotten-Baumann. Los haluros de ácido se hidrolizan en presencia de agua para producir ácidos carboxílicos, pero este tipo de reacción rara vez es útil, ya que los ácidos carboxílicos se usan típicamente para sintetizar haluros de ácido. La mayoría de las reacciones con haluros de ácido se llevan a cabo en presencia de una base no nucleófila, como la piridina, para neutralizar el ácido hidrohálico que se forma como subproducto.
Los haluros de ácido reaccionarán con nucleófilos de carbono, como Grignards y enolatos, aunque pueden resultar mezclas de productos. Si bien un nucleófilo de carbono reaccionará primero con el haluro de ácido para producir una cetona, la cetona también es susceptible al ataque nucleófilo y puede convertirse en un alcohol terciario. Por ejemplo, cuando el cloruro de benzoílo (1) se trata con dos equivalentes de un reactivo de Grignard, como bromuro de metilmagnesio (MeMgBr), se obtiene 2-fenil-2-propanol (3) con un rendimiento excelente. Aunque la acetofenona (2) es un intermedio en esta reacción, es imposible aislarla porque reacciona con un segundo equivalente de MeMgBr rápidamente después de formarse.

A diferencia de la mayoría de los otros nucleófilos de carbono, los dialquilcupratos de litio, a menudo llamados reactivos de Gilman, pueden agregarse a los haluros de ácido solo una vez para dar cetonas. Sin embargo, la reacción entre un haluro de ácido y un reactivo de Gilman no es una reacción de sustitución de acilo nucleofílica y se cree que procede a través de una vía de radicales. La síntesis de cetonas de Weinreb también se puede utilizar para convertir haluros de ácido en cetonas. En esta reacción, el haluro de ácido se convierte primero en una N-metoxi-N-metilamida, conocida como amida de Weinreb. Cuando un nucleófilo de carbono, como un reactivo de Grignard o de organolitio, se agrega a una amida de Weinreb, el metal es quelado por los oxígenos carbonilo y N-metoxi, lo que evita nuevas adiciones nucleófilas.
En la acilación de Friedel-Crafts, los haluros de ácido actúan como electrófilos para la sustitución aromática electrófila. Un ácido de Lewis, como el cloruro de zinc (ZnCl 2), el cloruro de hierro (III) (FeCl 3) o el cloruro de aluminio (AlCl 3), se coordina con el halógeno en el haluro de ácido, activando el compuesto hacia el ataque nucleofílico por un aromático activado. anillo. Para anillos aromáticos especialmente ricos en electrones, la reacción procederá sin un ácido de Lewis.
Tioésteres
La química de los tioésteres y los haluros de ácido es similar, la reactividad recuerda a la de los cloruros de ácido, pero es más suave.
Anhídridos
La química de los haluros y anhídridos de ácido es similar. Si bien los anhídridos no se pueden convertir en haluros de ácido, se pueden convertir en los derivados de acilo restantes. Los anhídridos también participan en reacciones tipo Schotten-Baumann para producir ésteres y amidas a partir de alcoholes y aminas, y el agua puede hidrolizar los anhídridos a sus correspondientes ácidos. Al igual que los haluros de ácido, los anhídridos también pueden reaccionar con nucleófilos de carbono para producir cetonas y/o alcoholes terciarios, y pueden participar tanto en la acilación de Friedel-Crafts como en la síntesis de cetonas de Weinreb. Sin embargo, a diferencia de los haluros de ácido, los anhídridos no reaccionan con los reactivos de Gilman.
La reactividad de los anhídridos se puede aumentar usando una cantidad catalítica de N,N-dimetilaminopiridina o DMAP. La piridina también se puede usar para este propósito y actúa a través de un mecanismo similar.

Primero, DMAP (2) ataca al anhídrido (1) para formar un intermedio tetraédrico, que colapsa para eliminar un ion carboxilato para dar la amida 3. Esta amida intermedia está más activada hacia el ataque nucleofílico que el anhídrido original, porque la dimetilaminopiridina es un mejor grupo saliente que un carboxilato. En el conjunto final de pasos, un nucleófilo (Nuc) ataca a 3 para dar otro intermedio tetraédrico. Cuando este intermedio colapsa para dar el producto 4, el grupo piridina se elimina y se restaura su aromaticidad, una poderosa fuerza impulsora y la razón por la cual el compuesto de piridina es un mejor grupo saliente que un ion carboxilato.
ésteres
Los ésteres son menos reactivos que los haluros y anhídridos de ácido. Al igual que con los derivados de acilo más reactivos, pueden reaccionar con amoníaco y aminas primarias y secundarias para dar amidas, aunque este tipo de reacción no se usa con frecuencia, ya que los haluros de ácido dan mejores rendimientos. Los ésteres se pueden convertir en otros ésteres en un proceso conocido como transesterificación. La transesterificación puede ser catalizada por ácidos o bases, e implica la reacción de un éster con un alcohol. Desafortunadamente, debido a que el grupo saliente también es un alcohol, las reacciones directa e inversa a menudo ocurrirán a velocidades similares. El uso de un gran exceso del alcohol reactivo o la eliminación del alcohol del grupo saliente (p. ej., mediante destilación) impulsará la reacción hacia la finalización, de acuerdo con el principio de Le Chatelier.
La hidrólisis de ésteres catalizada por ácido también es un proceso de equilibrio, esencialmente lo contrario de la reacción de esterificación de Fischer. Debido a que un alcohol (que actúa como grupo saliente) y el agua (que actúa como nucleófilo) tienen valores p K a similares, las reacciones directa e inversa compiten entre sí. Al igual que en la transesterificación, el uso de un gran exceso de reactivo (agua) o la eliminación de uno de los productos (el alcohol) puede promover la reacción directa.
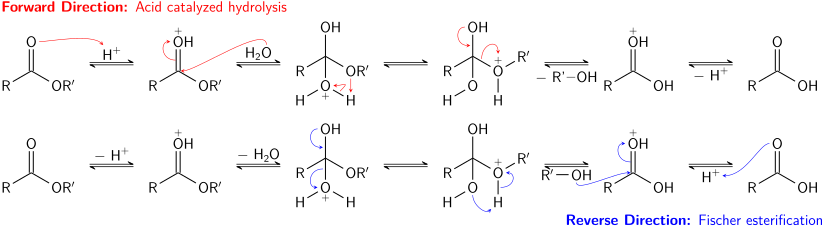
La hidrólisis básica de ésteres, conocida como saponificación, no es un proceso de equilibrio; en la reacción se consume un equivalente completo de base, lo que produce un equivalente de alcohol y un equivalente de una sal de carboxilato. La saponificación de ésteres de ácidos grasos es un proceso industrialmente importante, utilizado en la producción de jabón.
Los ésteres pueden experimentar una variedad de reacciones con nucleófilos de carbono. Al igual que los haluros y anhídridos de ácido, reaccionarán con un exceso de reactivo de Grignard para dar alcoholes terciarios. Los ésteres también reaccionan fácilmente con los enolatos. En la condensación de Claisen, un enolato de un éster (1) atacará al grupo carbonilo de otro éster (2) para dar el intermedio tetraédrico 3. El intermedio colapsa, expulsando un alcóxido (R'O) y produciendo β-cetoéster 4.

También son posibles las condensaciones de Claisen cruzadas, en las que el enolato y el nucleófilo son ésteres diferentes. Una condensación de Claisen intramolecular se denomina condensación de Dieckmann o ciclación de Dieckmann, ya que se puede utilizar para formar anillos. Los ésteres también pueden sufrir condensaciones con enolatos de cetonas y aldehídos para dar compuestos de β-dicarbonilo. Un ejemplo específico de esto es el reordenamiento de Baker-Venkataraman, en el que una ortoaciloxicetona aromática sufre una sustitución de acilo nucleofílica intramolecular y un reordenamiento posterior para formar una β-dicetona aromática. El reordenamiento de Chan es otro ejemplo de un reordenamiento resultante de una reacción de sustitución de acilo nucleofílica intramolecular.
Amidas
Debido a su baja reactividad, las amidas no participan en tantas reacciones de sustitución nucleófila como lo hacen otros derivados de acilo. Las amidas son estables al agua y son aproximadamente 100 veces más estables a la hidrólisis que los ésteres. Sin embargo, las amidas pueden hidrolizarse a ácidos carboxílicos en presencia de ácido o base. La estabilidad de los enlaces amida tiene implicaciones biológicas, ya que los aminoácidos que componen las proteínas están enlazados con enlaces amida. Los enlaces amida son lo suficientemente resistentes a la hidrólisis para mantener la estructura de la proteína en ambientes acuosos, pero son lo suficientemente susceptibles como para romperse cuando sea necesario.
Las amidas primarias y secundarias no reaccionan favorablemente con los nucleófilos de carbono. Los reactivos de Grignard y los organolitios actuarán como bases en lugar de nucleófilos y simplemente desprotonarán la amida. Las amidas terciarias no experimentan este problema y reaccionan con nucleófilos de carbono para dar cetonas; el anión amida (NR 2) es una base muy fuerte y, por lo tanto, un grupo saliente muy pobre, por lo que el ataque nucleofílico solo ocurre una vez. Cuando se hace reaccionar con nucleófilos de carbono, se puede usar N, N -dimetilformamida (DMF) para introducir un grupo formilo.

Aquí, el fenillitio 1 ataca al grupo carbonilo de DMF 2, dando el intermedio tetraédrico 3. Debido a que el anión dimetilamida es un grupo saliente pobre, el intermedio no colapsa y no ocurre otra adición nucleófila. Tras el tratamiento ácido, el alcóxido se protona para dar 4, luego la amina se protona para dar 5. La eliminación de una molécula neutra de dimetilamina y la pérdida de un protón dan benzaldehído, 6.
Ácidos carboxílicos
Los ácidos carboxílicos no son especialmente reactivos frente a la sustitución nucleófila, aunque pueden convertirse en otros derivados de acilo. Es posible convertir un ácido carboxílico en una amida, pero no es sencillo. En lugar de actuar como nucleófilo, una amina reaccionará como base en presencia de un ácido carboxílico para dar la sal de carboxilato de amonio. Calentar la sal por encima de 100 °C eliminará el agua y conducirá a la formación de la amida. Este método de síntesis de amidas es industrialmente importante y también tiene aplicaciones de laboratorio.En presencia de un catalizador ácido fuerte, los ácidos carboxílicos pueden condensarse para formar anhídridos de ácido. Sin embargo, la condensación produce agua, que puede hidrolizar el anhídrido de nuevo a los ácidos carboxílicos iniciales. Así, la formación del anhídrido por condensación es un proceso de equilibrio.
En condiciones catalizadas por ácidos, los ácidos carboxílicos reaccionarán con los alcoholes para formar ésteres a través de la reacción de esterificación de Fischer, que también es un proceso de equilibrio. Alternativamente, se puede usar diazometano para convertir un ácido en un éster. Si bien las reacciones de esterificación con diazometano a menudo dan rendimientos cuantitativos, el diazometano solo es útil para formar ésteres metílicos.
El cloruro de tionilo se puede utilizar para convertir ácidos carboxílicos en sus correspondientes cloruros de acilo. Primero, el ácido carboxílico 1 ataca el cloruro de tionilo y el ion cloruro se va. El ion oxonio 2 resultante se activa hacia el ataque nucleofílico y tiene un buen grupo saliente, lo que lo distingue de un ácido carboxílico normal. En el siguiente paso, 2 es atacado por iones de cloruro para dar el intermedio tetraédrico 3, un clorosulfito. El intermedio tetraédrico colapsa con la pérdida de dióxido de azufre y iones de cloruro, dando cloruro de acilo protonado 4. El ion cloruro puede eliminar el protón del grupo carbonilo, dando el cloruro de acilo 5 con una pérdida de HCl.

El cloruro de fósforo (III) (PCl 3) y el cloruro de fósforo (V) (PCl 5) también convertirán los ácidos carboxílicos en cloruros de ácido mediante un mecanismo similar. Un equivalente de PCl 3 puede reaccionar con tres equivalentes de ácido, produciendo un equivalente de H 3 PO 3, o ácido fosforoso, además del cloruro de ácido deseado. El PCl 5 reacciona con los ácidos carboxílicos en una proporción de 1:1 y produce oxicloruro de fósforo (V) (POCl 3) y cloruro de hidrógeno (HCl) como subproductos.
Los ácidos carboxílicos reaccionan con los reactivos de Grignard y los organolitios para formar cetonas. El primer equivalente de nucleófilo actúa como base y desprotona el ácido. Un segundo equivalente atacará al grupo carbonilo para crear un dianión de alcóxido geminal, que se protona tras el procesamiento para dar el hidrato de una cetona. Debido a que la mayoría de los hidratos de cetonas son inestables en relación con sus cetonas correspondientes, el equilibrio entre los dos se desplaza fuertemente a favor de la cetona. Por ejemplo, la constante de equilibrio para la formación de hidrato de acetona a partir de acetona es solo 0,002. El grupo carboxílico es el más ácido de los compuestos orgánicos.
Contenido relacionado
Tacticidad
Electrón de valencia
Actina