
Síntesis enantioselectiva
La síntesis enantioselectiva, también llamada síntesis asimétrica, es una forma de síntesis química. La IUPAC la define como "una reacción química (o secuencia de reacción) en la que se forman uno o más elementos nuevos de quiralidad en una molécula de sustrato y que produce productos estereoisómeros (enantioméricos o diastereoisómeros) en cantidades desiguales".
Dicho de manera más simple: es la síntesis de un compuesto por un método que favorece la formación de un enantiómero o diastereoisómero específico. Los enantiómeros son estereoisómeros que tienen configuraciones opuestas en cada centro quiral. Los diastereómeros son estereoisómeros que difieren en uno o más centros quirales.
La síntesis enantioselectiva es un proceso clave en la química moderna y es particularmente importante en el campo de los productos farmacéuticos, ya que los diferentes enantiómeros o diastereómeros de una molécula a menudo tienen una actividad biológica diferente.
Visión general
Muchos de los componentes básicos de los sistemas biológicos, como los azúcares y los aminoácidos, se producen exclusivamente como un enantiómero. Como resultado, los sistemas vivos poseen un alto grado de quiralidad química y, a menudo, reaccionarán de manera diferente con los diversos enantiómeros de un compuesto dado. Ejemplos de esta selectividad incluyen:
- Sabor: el edulcorante artificial aspartamo tiene dos enantiómeros. El L -aspartamo tiene un sabor dulce mientras que el D -aspartamo no tiene sabor.
- Olor: R -(–)-carvona huele a menta verde mientras que S -(+)-carvona huele a alcaravea.
- Eficacia del medicamento: el medicamento antidepresivo Citalopram se vende como una mezcla racémica. Sin embargo, los estudios han demostrado que solo el enantiómero (S)-(+) es responsable de los efectos beneficiosos del fármaco.
- Seguridad del fármaco: la D -penicilamina se utiliza en la terapia de quelación y para el tratamiento de la artritis reumatoide, mientras que la L -penicilamina es tóxica porque inhibe la acción de la piridoxina, una vitamina B esencial.
Como tal, la síntesis enantioselectiva es de gran importancia pero también puede ser difícil de lograr. Los enantiómeros poseen entalpías y entropías idénticas y, por lo tanto, deben producirse en cantidades iguales mediante un proceso no dirigido, lo que lleva a una mezcla racémica. La síntesis enantioselectiva se puede lograr utilizando una característica quiral que favorece la formación de un enantiómero sobre otro a través de interacciones en el estado de transición. Esta polarización se conoce como inducción asimétrica y puede involucrar características quirales en el sustrato, el reactivo, el catalizador o el entorno y funciona haciendo que la energía de activación requerida para formar un enantiómero sea más baja que la del enantiómero opuesto.
La enantioselectividad generalmente está determinada por las tasas relativas de un paso de diferenciación enantiomérica, el punto en el que un reactivo puede convertirse en cualquiera de los dos productos enantioméricos. La constante de velocidad, k, para una reacción es función de la energía de activación de la reacción, a veces denominada barrera de energía, y depende de la temperatura. Usando la energía libre de Gibbs de la barrera de energía, Δ G *, significa que las tasas relativas para resultados estereoquímicos opuestos a una temperatura dada, T, es:

Esta dependencia de la temperatura significa que la diferencia de velocidad, y por lo tanto la enantioselectividad, es mayor a temperaturas más bajas. Como resultado, incluso las pequeñas diferencias en la barrera de energía pueden generar un efecto notable.
| ΔΔ G * (kcal) | k 1/k 2a 273 K | k 1/k 2a 298 K | k 1/k 2a 323K) | |||
|---|---|---|---|---|---|---|
| 1.0 | 6 | .37 | 5 | .46 | 4 | .78 |
| 2.0 | 40 | .6 | 29 | .8 | 22 | .9 |
| 3.0 | 259 | 162 | 109 | |||
| 4.0 | 1650 | 886 | 524 | |||
| 5.0 | 10500 | 4830 | 2510 |
Enfoques
Catálisis enantioselectiva
La catálisis enantioselectiva (conocida tradicionalmente como "catálisis asimétrica") se realiza utilizando catalizadores quirales, que suelen ser complejos de coordinación quirales. La catálisis es eficaz para una gama más amplia de transformaciones que cualquier otro método de síntesis enantioselectiva. Los catalizadores metálicos quirales se vuelven casi invariablemente quirales mediante el uso de ligandos quirales, pero es posible generar complejos quirales en metal compuestos en su totalidad por ligandos aquirales. La mayoría de los catalizadores enantioselectivos son efectivos en proporciones bajas de sustrato/catalizador. Dadas sus altas eficiencias, a menudo son adecuados para la síntesis a escala industrial, incluso con catalizadores caros. Un ejemplo versátil de síntesis enantioselectiva es la hidrogenación asimétrica, que se utiliza para reducir una amplia variedad de grupos funcionales.

El diseño de nuevos catalizadores está dominado por el desarrollo de nuevas clases de ligandos. Ciertos ligandos, a menudo denominados "ligandos privilegiados", son efectivos en una amplia gama de reacciones; los ejemplos incluyen BINOL, Salen y BOX. La mayoría de los catalizadores son efectivos para un solo tipo de reacción asimétrica. Por ejemplo, la hidrogenación asimétrica de Noyori con BINAP/Ru requiere una β-cetona, aunque otro catalizador, BINAP/diamina-Ru, amplía el alcance a α, β-alquenos y productos químicos aromáticos.
Auxiliares quirales
Un auxiliar quiral es un compuesto orgánico que se acopla al material de partida para formar un nuevo compuesto que luego puede sufrir reacciones diastereoselectivas a través de la inducción asimétrica intramolecular. Al final de la reacción, se elimina el auxiliar, en condiciones que no provocarán la racemización del producto. Por lo general, luego se recupera para uso futuro.

Los auxiliares quirales deben usarse en cantidades estequiométricas para que sean efectivos y requieren pasos sintéticos adicionales para añadir y eliminar el auxiliar. Sin embargo, en algunos casos, la única metodología estereoselectiva disponible se basa en auxiliares quirales y estas reacciones tienden a ser versátiles y muy bien estudiadas, lo que permite el acceso más eficiente en el tiempo a productos enantioméricamente puros. Además, los productos de las reacciones dirigidas por auxiliares son diastereómeros, lo que permite su fácil separación mediante métodos como la cromatografía en columna o la cristalización.
Biocatálisis
La biocatálisis utiliza compuestos biológicos, que van desde enzimas aisladas hasta células vivas, para realizar transformaciones químicas. Las ventajas de estos reactivos incluyen un ee muy alto y una especificidad del reactivo, así como condiciones de operación suaves y un bajo impacto ambiental. Los biocatalizadores se usan más comúnmente en la industria que en la investigación académica; por ejemplo en la producción de estatinas. Sin embargo, la alta especificidad del reactivo puede ser un problema, ya que a menudo requiere que se evalúe una amplia gama de biocatalizadores antes de encontrar un reactivo eficaz.
Organocatálisis enantioselectiva
La organocatálisis se refiere a una forma de catálisis en la que la velocidad de una reacción química aumenta mediante un compuesto orgánico que consta de carbono, hidrógeno, azufre y otros elementos no metálicos. Cuando el organocatalizador es quiral, se puede lograr la síntesis enantioselectiva; por ejemplo, varias reacciones de formación de enlaces carbono-carbono se vuelven enantioselectivas en presencia de prolina, siendo la reacción aldólica un buen ejemplo. La organocatálisis a menudo emplea compuestos naturales y aminas secundarias como catalizadores quirales; estos son económicos y respetuosos con el medio ambiente, ya que no se utilizan metales.
Síntesis de grupos quirales
La síntesis de grupos quirales es uno de los enfoques más simples y antiguos para la síntesis enantioselectiva. Un material de partida quiral fácilmente disponible se manipula a través de reacciones sucesivas, a menudo utilizando reactivos aquirales, para obtener la molécula objetivo deseada. Esto puede cumplir los criterios para la síntesis enantioselectiva cuando se crea una nueva especie quiral, como en una reacción S N 2.

La síntesis de grupos quirales es especialmente atractiva para las moléculas objetivo que tienen una quiralidad similar a un bloque de construcción natural relativamente económico, como un azúcar o un aminoácido. Sin embargo, el número de posibles reacciones que puede experimentar la molécula está restringido y pueden requerirse rutas de síntesis tortuosas (p. ej., síntesis total de oseltamivir). Este enfoque también requiere una cantidad estequiométrica del material de partida enantiopuro, que puede ser costoso si no se produce de forma natural.
Separación y análisis de enantiómeros
Los dos enantiómeros de una molécula poseen las mismas propiedades físicas (por ejemplo, punto de fusión, punto de ebullición, polaridad, etc.) y, por tanto, se comportan de forma idéntica entre sí. Como resultado, migrarán con un Rf idéntico en cromatografía de capa fina y tendrán tiempos de retención idénticos en HPLC y GC. Sus espectros de RMN e IR son idénticos.
Esto puede hacer que sea muy difícil determinar si un proceso ha producido un solo enantiómero (y, lo que es más importante, qué enantiómero es), además de dificultar la separación de los enantiómeros de una reacción que no ha sido 100 % enantioselectiva. Afortunadamente, los enantiómeros se comportan de manera diferente en presencia de otros materiales quirales y esto puede aprovecharse para permitir su separación y análisis.
Los enantiómeros no migran de forma idéntica en los medios cromatográficos quirales, como el cuarzo o los medios estándar modificados quiralmente. Esto constituye la base de la cromatografía en columna quiral, que se puede utilizar a pequeña escala para permitir el análisis mediante GC y HPLC, oa gran escala para separar materiales quiralmente impuros. Sin embargo, este proceso puede requerir una gran cantidad de material de relleno quiral que puede ser costoso. Una alternativa común es usar un agente de derivación quiral para convertir los enantiómeros en diastereómeros, de la misma manera que los auxiliares quirales. Estos tienen diferentes propiedades físicas y, por lo tanto, pueden separarse y analizarse utilizando métodos convencionales. Los agentes especiales de derivatización quiral conocidos como "agentes de resolución quiral" se utilizan en la espectroscopia de RMN de estereoisómeros,3 y Eu(hfc) 3.
La separación y el análisis de los enantiómeros componentes de fármacos racémicos o sustancias farmacéuticas se conocen como análisis quiral. o análisis enantioselectivo. La técnica empleada con más frecuencia para llevar a cabo el análisis quiral involucra procedimientos de ciencia de separación, específicamente métodos cromatográficos quirales.
El exceso enantiomérico de una sustancia también se puede determinar utilizando ciertos métodos ópticos. El método más antiguo para hacer esto es usar un polarímetro para comparar el nivel de rotación óptica en el producto contra un 'estándar' de composición conocida. También es posible realizar espectroscopia ultravioleta-visible de estereoisómeros explotando el efecto Cotton.
Una de las formas más precisas de determinar la quiralidad de un compuesto es determinar su configuración absoluta mediante cristalografía de rayos X. Sin embargo, este es un proceso laborioso que requiere que se cultive un monocristal adecuado.
Historia
Inicio (1815-1905)
En 1815, el físico francés Jean-Baptiste Biot demostró que ciertas sustancias químicas podían rotar el plano de un haz de luz polarizada, una propiedad denominada actividad óptica. La naturaleza de esta propiedad siguió siendo un misterio hasta 1848, cuando Louis Pasteur propuso que tenía una base molecular que se originaba en alguna forma de disimetría, y Lord Kelvin acuñó el término quiralidad un año después. El origen de la quiralidad en sí se describió finalmente en 1874, cuando Jacobus Henricus van 't Hoff y Joseph Le Bel propusieron de forma independiente la geometría tetraédrica del carbono.Los modelos estructurales anteriores a este trabajo habían sido bidimensionales, y van 't Hoff y Le Bel teorizaron que la disposición de los grupos alrededor de este tetraedro podría dictar la actividad óptica del compuesto resultante a través de lo que se conoció como Le Bel–van 't regla de Hoff.
En 1894 Hermann Emil Fischer esbozó el concepto de inducción asimétrica; en el que correctamente atribuyó selectivamente la formación de D -glucosa por las plantas debido a la influencia de sustancias ópticamente activas dentro de la clorofila. Fischer también realizó con éxito lo que ahora se consideraría como el primer ejemplo de síntesis enantioselectiva, mediante la elongación enantioselectiva de azúcares a través de un proceso que eventualmente se convertiría en la síntesis de Kiliani-Fischer.
La primera síntesis química enantioselectiva se atribuye con mayor frecuencia a Willy Marckwald, Universität zu Berlin, para una descarboxilación enantioselectiva catalizada por brucina del ácido 2-etil-2-metilmalónico informada en 1904. Un ligero exceso de la forma levógira del producto de la reacción, se produjo ácido 2-metilbutírico; como este producto también es un producto natural, por ejemplo, como una cadena lateral de lovastatina formada por su dicetide sintasa (LovF) durante su biosíntesis, este resultado constituye la primera síntesis total registrada con enantioselectividad, así como otras primicias (como señala Koskinen, primera "ejemplo de catálisis asimétrica, selección enantiotópica y organocatálisis").Esta observación también tiene un significado histórico, ya que en ese momento la síntesis enantioselectiva solo podía entenderse en términos de vitalismo. En ese momento, muchos químicos prominentes como Jöns Jacob Berzelius argumentaron que los compuestos naturales y artificiales eran fundamentalmente diferentes y que la quiralidad era simplemente una manifestación de la "fuerza vital" que solo podía existir en los compuestos naturales. A diferencia de Fischer, Marckwald había realizado una reacción enantioselectiva sobre un material de partida no natural aquiral, aunque con un organocatalizador quiral (como ahora entendemos esta química).
Primeros trabajos (1905-1965)
El desarrollo de la síntesis enantioselectiva fue inicialmente lento, en gran parte debido a la limitada gama de técnicas disponibles para su separación y análisis. Los diastereómeros poseen diferentes propiedades físicas, lo que permite la separación por medios convencionales; sin embargo, en ese momento, los enantiómeros solo podían separarse por resolución espontánea (donde los enantiómeros se separan tras la cristalización) o resolución cinética (donde un enantiómero se destruye selectivamente). La única herramienta para analizar los enantiómeros fue la actividad óptica mediante un polarímetro, un método que no proporciona datos estructurales.
No fue sino hasta la década de 1950 que realmente comenzó un gran progreso. Impulsado en parte por químicos como RB Woodward y Vladimir Prelog, pero también por el desarrollo de nuevas técnicas. El primero de ellos fue la cristalografía de rayos X, que se utilizó para determinar la configuración absoluta de un compuesto orgánico por Johannes Bijvoet en 1951. La cromatografía quiral fue introducida un año después por Dalgliesh, quien utilizó la cromatografía en papel para separar los aminoácidos quirales. Aunque Dalgliesh no fue el primero en observar tales separaciones, atribuyó correctamente la separación de enantiómeros a la retención diferencial por parte de la celulosa quiral. Esto se amplió en 1960, cuando Klem y Reed informaron por primera vez sobre el uso de gel de sílice modificado quiralmente para la separación por HPLC quiral.
Talidomida
Si bien se sabía que los diferentes enantiómeros de un fármaco podían tener diferentes actividades, Arthur Robertson Cushny realizó un importante trabajo inicial, pero esto no se tuvo en cuenta en el diseño y las pruebas iniciales del fármaco. Sin embargo, tras el desastre de la talidomida, el desarrollo y la concesión de licencias de fármacos cambiaron drásticamente.
Sintetizada por primera vez en 1953, la talidomida se prescribió ampliamente para las náuseas matutinas entre 1957 y 1962, pero pronto se descubrió que era gravemente teratogénica y eventualmente causó defectos de nacimiento en más de 10,000 bebés. El desastre llevó a muchos países a introducir normas más estrictas para las pruebas y la autorización de medicamentos, como la Enmienda Kefauver-Harris (EE. UU.) y la Directiva 65/65/EEC1 (UE).
Las primeras investigaciones sobre el mecanismo teratogénico, utilizando ratones, sugirieron que un enantiómero de la talidomida era teratogénico mientras que el otro poseía toda la actividad terapéutica. Más tarde se demostró que esta teoría era incorrecta y ahora ha sido reemplazada por un cuerpo de investigación. Sin embargo, aumentó la importancia de la quiralidad en el diseño de fármacos, lo que llevó a una mayor investigación sobre la síntesis enantioselectiva.
Edad moderna (desde 1965)
Las reglas de prioridad de Cahn-Ingold-Prelog (a menudo abreviadas como el sistema CIP) se publicaron por primera vez en 1966; permitiendo que los enantiómeros se describan con mayor facilidad y precisión. El mismo año vio la primera separación enantiomérica exitosa por cromatografía de gases, un desarrollo importante ya que la tecnología era de uso común en ese momento.
La síntesis enantioselectiva catalizada por metales fue iniciada por William S. Knowles, Ryōji Noyori y K. Barry Sharpless; por lo que recibirían el Premio Nobel de Química 2001. Knowles y Noyori comenzaron con el desarrollo de la hidrogenación asimétrica, que desarrollaron de forma independiente en 1968. Knowles reemplazó los ligandos de trifenilfosfina aquirales en el catalizador de Wilkinson con ligandos de fosfina quirales. Este catalizador experimental se empleó en una hidrogenación asimétrica con un modesto exceso enantiomérico del 15%. Knowles también fue el primero en aplicar la catálisis enantioselectiva de metales a la síntesis a escala industrial; mientras trabajaba para Monsanto Company, desarrolló un paso de hidrogenación enantioselectiva para la producción de L-DOPA, utilizando el ligando DIPAMP.
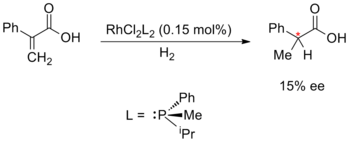 | 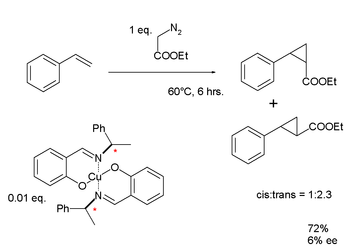 | |
| Knowles: hidrogenación asimétrica (1968) | Noyori: ciclopropanación enantioselectiva (1968) |
|---|
Noyori ideó un complejo de cobre utilizando un ligando base de Schiff quiral, que utilizó para la ciclopropanación de estireno con metal-carbenoide. Al igual que los hallazgos de Knowles, los resultados de Noyori para el exceso enantiomérico de este ligando de primera generación fueron decepcionantemente bajos: 6 %. Sin embargo, la investigación continua finalmente condujo al desarrollo de la reacción de hidrogenación asimétrica de Noyori.
Sharpless complementó estas reacciones de reducción mediante el desarrollo de una gama de oxidaciones asimétricas (epoxidación de Sharpless, dihidroxilación asimétrica de Sharpless, oxiaminación de Sharpless) durante las décadas de 1970 y 1980. Siendo la reacción de oxiaminación asimétrica, utilizando tetróxido de osmio, la más temprana.
Durante el mismo período, se desarrollaron métodos para permitir el análisis de compuestos quirales por RMN; ya sea usando agentes derivatizantes quirales, como el ácido de Mosher, o reactivos de desplazamiento a base de europio, de los cuales Eu(DPM) 3 fue el primero.
Los auxiliares quirales fueron introducidos por EJ Corey en 1978 y ocuparon un lugar destacado en el trabajo de Dieter Enders. Casi al mismo tiempo se desarrolló la organocatálisis enantioselectiva, con trabajos pioneros que incluyen la reacción de Hajos-Parrish-Eder-Sauer-Wiechert. Las reacciones enantioselectivas catalizadas por enzimas se hicieron cada vez más comunes durante la década de 1980, particularmente en la industria, y sus aplicaciones incluían la hidrólisis asimétrica de ésteres con esterasa de hígado de cerdo. La tecnología emergente de la ingeniería genética ha permitido la adaptación de enzimas a procesos específicos, lo que permite una mayor variedad de transformaciones selectivas. Por ejemplo, en la hidrogenación asimétrica de precursores de estatinas.
Contenido relacionado
Quimiosíntesis
Coenzima Q – citocromo c reductasa
Claude Louis Berthollet