
Reacción en cadena de la polimerasa (PCR)
La reacción en cadena de la polimerasa (PCR por sus siglas del inglés polymerase chain reaction) es un método ampliamente utilizado para hacer rápidamente de millones a miles de millones de copias (completas o parciales) de una muestra específica de ADN, lo que permite a los científicos tomar una muestra muy pequeña de ADN y amplificarla (o una parte de ella).) a una cantidad lo suficientemente grande como para estudiar en detalle. PCR fue inventado en 1983 por el bioquímico estadounidense Kary Mullis en Cetus Corporation; Mullis y el bioquímico Michael Smith, que habían desarrollado otras formas esenciales de manipular el ADN, recibieron conjuntamente el Premio Nobel de Química en 1993.
La PCR es fundamental para muchos de los procedimientos utilizados en las pruebas e investigaciones genéticas, incluido el análisis de muestras antiguas de ADN y la identificación de agentes infecciosos. Usando PCR, las copias de cantidades muy pequeñas de secuencias de ADN se amplifican exponencialmente en una serie de ciclos de cambios de temperatura. La PCR es ahora una técnica común y, a menudo, indispensable que se utiliza en la investigación de laboratorio médico para una amplia variedad de aplicaciones, incluida la investigación biomédica y la ciencia forense criminal.
La mayoría de los métodos de PCR se basan en ciclos térmicos. El ciclo térmico expone a los reactivos a ciclos repetidos de calentamiento y enfriamiento para permitir diferentes reacciones dependientes de la temperatura, específicamente, la fusión del ADN y la replicación del ADN impulsada por enzimas. La PCR emplea dos reactivos principales: cebadores (que son fragmentos cortos de ADN de cadena sencilla conocidos como oligonucleótidos que son una secuencia complementaria a la región de ADN objetivo) y una ADN polimerasa. En el primer paso de la PCR, las dos hebras de la doble hélice del ADN se separan físicamente a alta temperatura en un proceso llamado desnaturalización del ácido nucleico. En el segundo paso, se baja la temperatura y los cebadores se unen a las secuencias complementarias de ADN. Luego, las dos cadenas de ADN se convierten en plantillas para que la ADN polimerasa ensamble enzimáticamente una nueva cadena de ADN a partir de nucleótidos libres, los componentes básicos del ADN.
Casi todas las aplicaciones de PCR emplean una polimerasa de ADN estable al calor, como la polimerasa Taq, una enzima aislada originalmente de la bacteria termófila Thermus aquaticus. Si la polimerasa usada fuera sensible al calor, se desnaturalizaría bajo las altas temperaturas del paso de desnaturalización. Antes del uso de la polimerasa Taq, la polimerasa de ADN tenía que agregarse manualmente en cada ciclo, lo que era un proceso tedioso y costoso.
Las aplicaciones de la técnica incluyen la clonación de ADN para la secuenciación, la clonación y manipulación de genes, la mutagénesis de genes; construcción de filogenias basadas en ADN o análisis funcional de genes; diagnóstico y seguimiento de trastornos genéticos; amplificación de ADN antiguo; análisis de huellas dactilares genéticas para perfiles de ADN (por ejemplo, en ciencia forense y pruebas de paternidad); y detección de patógenos en pruebas de ácido nucleico para el diagnóstico de enfermedades infecciosas.
Principios
La PCR amplifica una región específica de una cadena de ADN (el objetivo de ADN). La mayoría de los métodos de PCR amplifican fragmentos de ADN de entre 0,1 y 10 kilopares de bases (kpb) de longitud, aunque algunas técnicas permiten la amplificación de fragmentos de hasta 40 kpb. La cantidad de producto amplificado está determinada por los sustratos disponibles en la reacción, que se vuelve limitante a medida que avanza la reacción.
Una configuración básica de PCR requiere varios componentes y reactivos, que incluyen:
- una plantilla de ADN que contiene la región objetivo de ADN para amplificar
- una ADN polimerasa; una enzima que polimeriza nuevas hebras de ADN; La polimerasa Taq resistente al calor es especialmente común, ya que es más probable que permanezca intacta durante el proceso de desnaturalización del ADN a alta temperatura.
- dos cebadores de ADN que son complementarios a los extremos 3' (tres cebadores) de cada una de las cadenas con sentido y antisentido del objetivo de ADN (la ADN polimerasa solo puede unirse y alargarse de una región de ADN de doble cadena; sin cebadores, no hay un sitio de iniciación de doble cadena en el que la polimerasa pueda unirse); Los cebadores específicos que son complementarios a la región objetivo del ADN se seleccionan de antemano y, a menudo, se fabrican a medida en un laboratorio o se compran a proveedores bioquímicos comerciales.
- trifosfatos de desoxinucleósidos, o dNTP (a veces llamados "trifosfatos de desoxinucleótidos"; nucleótidos que contienen grupos trifosfato), los componentes básicos a partir de los cuales la ADN polimerasa sintetiza una nueva hebra de ADN
- una solución tampón que proporciona un entorno químico adecuado para una actividad y estabilidad óptimas de la ADN polimerasa
- cationes bivalentes, típicamente iones de magnesio (Mg) o manganeso (Mn); Mg es el más común, pero Mn se puede usar para la mutagénesis de ADN mediada por PCR, ya que una concentración más alta de Mn aumenta la tasa de error durante la síntesis de ADN; y cationes monovalentes, típicamente iones de potasio (K)
La reacción suele llevarse a cabo en un volumen de 10 a 200 μl en pequeños tubos de reacción (volúmenes de 0,2 a 0,5 ml) en un termociclador. El termociclador calienta y enfría los tubos de reacción para alcanzar las temperaturas requeridas en cada paso de la reacción (ver más abajo). Muchos termocicladores modernos utilizan el efecto Peltier, que permite calentar y enfriar el bloque que contiene los tubos de PCR simplemente invirtiendo la corriente eléctrica. Los tubos de reacción de paredes delgadas permiten una conductividad térmica favorable para permitir un rápido equilibrio térmico. La mayoría de los termocicladores tienen tapas calentadas para evitar la condensación en la parte superior del tubo de reacción. Los termocicladores más antiguos que carecen de una tapa calentada requieren una capa de aceite encima de la mezcla de reacción o una bola de cera dentro del tubo.
Procedimiento
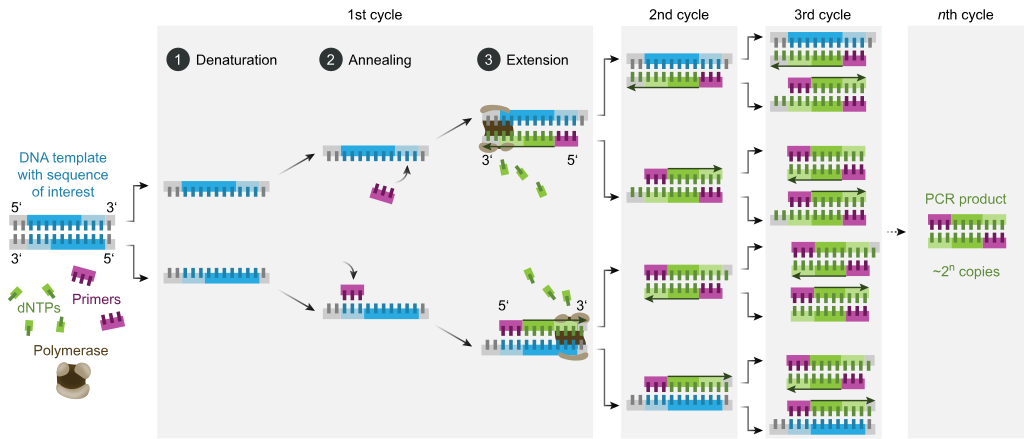
Por lo general, la PCR consiste en una serie de 20 a 40 cambios de temperatura repetidos, llamados ciclos térmicos, y cada ciclo consta comúnmente de dos o tres pasos de temperatura discretos (consulte la figura a continuación). El ciclo suele estar precedido por un solo paso de temperatura a una temperatura muy alta (>90 °C (194 °F)) y seguido por una retención al final para la extensión del producto final o almacenamiento breve. Las temperaturas utilizadas y el tiempo que se aplican en cada ciclo dependen de una variedad de parámetros, incluida la enzima utilizada para la síntesis de ADN, la concentración de iones bivalentes y dNTP en la reacción, y la temperatura de fusión (T m) de la cebadores Los pasos individuales comunes a la mayoría de los métodos de PCR son los siguientes:
- Inicialización: este paso solo se requiere para las ADN polimerasas que requieren activación por calor mediante PCR de inicio en caliente. Consiste en calentar la cámara de reacción a una temperatura de 94 a 96 °C (201 a 205 °F), o 98 °C (208 °F) si se utilizan polimerasas extremadamente termoestables, que luego se mantiene durante 1 a 10 minutos.
- Desnaturalización: este paso es el primer evento de ciclo regular y consiste en calentar la cámara de reacción a 94–98 °C (201–208 °F) durante 20–30 segundos. Esto provoca la fusión o desnaturalización del ADN de la plantilla de ADN de doble cadena al romper los enlaces de hidrógeno entre las bases complementarias, lo que produce dos moléculas de ADN de cadena sencilla.
- Hibridación: en el siguiente paso, la temperatura de reacción se reduce a 50–65 °C (122–149 °F) durante 20–40 segundos, lo que permite la hibridación de los cebadores con cada una de las plantillas de ADN monocatenario. Normalmente se incluyen dos cebadores diferentes en la mezcla de reacción: uno para cada uno de los dos complementos monocatenarios que contienen la región objetivo. Los cebadores son secuencias monocatenarias en sí mismas, pero son mucho más cortas que la longitud de la región objetivo y complementan solo secuencias muy cortas en el extremo 3' de cada cadena.
Es fundamental determinar una temperatura adecuada para el paso de recocido porque la eficiencia y la especificidad se ven fuertemente afectadas por la temperatura de recocido. Esta temperatura debe ser lo suficientemente baja para permitir la hibridación del cebador con la hebra, pero lo suficientemente alta para que la hibridación sea específica, es decir, el cebador debe unirse sólo a una parte perfectamente complementaria de la hebra y a ningún otro lugar. Si la temperatura es demasiado baja, la imprimación puede adherirse de manera imperfecta. Si es demasiado alto, es posible que la imprimación no se adhiera en absoluto. Una temperatura típica de recocido está entre 3 y 5 °C por debajo de la T mde las imprimaciones utilizadas. Los enlaces de hidrógeno estables entre bases complementarias se forman solo cuando la secuencia del cebador coincide muy de cerca con la secuencia de la plantilla. Durante este paso, la polimerasa se une al híbrido cebador-plantilla y comienza la formación de ADN.
- Extensión/alargamiento: La temperatura en este paso depende de la ADN polimerasa utilizada; la temperatura de actividad óptima para la ADN polimerasa termoestable de la polimerasa Taq es de aproximadamente 75 a 80 °C (167 a 176 °F),aunque comúnmente se usa una temperatura de 72 ° C (162 ° F) con esta enzima. En este paso, la ADN polimerasa sintetiza una nueva hebra de ADN complementaria a la hebra de la plantilla de ADN mediante la adición de dNTP libres de la mezcla de reacción que es complementaria a la plantilla en la dirección 5' a 3', condensando el grupo 5'-fosfato. de los dNTP con el grupo 3'-hidroxi al final de la cadena de ADN naciente (alargada). El tiempo preciso requerido para la elongación depende tanto de la ADN polimerasa utilizada como de la longitud de la región diana del ADN a amplificar. Como regla general, a su temperatura óptima, la mayoría de las ADN polimerasas polimerizan mil bases por minuto. En condiciones óptimas (es decir, si no hay limitaciones debidas a sustratos o reactivos limitantes), en cada paso de extensión/alargamiento, se duplica el número de secuencias diana de ADN.
Los procesos de desnaturalización, recocido y elongación constituyen un solo ciclo. Se requieren múltiples ciclos para amplificar el objetivo de ADN a millones de copias. La fórmula utilizada para calcular el número de copias de ADN formadas después de un número determinado de ciclos es 2, donde n es el número de ciclos. Por lo tanto, un conjunto de reacciones de 30 ciclos da como resultado 2, o 1.073.741.824, copias de la región diana del ADN de doble cadena original.
- Elongación final: este único paso es opcional, pero se realiza a una temperatura de 70 a 74 °C (158 a 165 °F) (el rango de temperatura requerido para una actividad óptima de la mayoría de las polimerasas utilizadas en PCR) durante 5 a 15 minutos después de la último ciclo de PCR para asegurarse de que cualquier resto de ADN monocatenario esté completamente elongado.
- Retención final: el paso final enfría la cámara de reacción a 4–15 °C (39–59 °F) durante un tiempo indefinido y puede emplearse para el almacenamiento a corto plazo de los productos de la PCR.
Para verificar si la PCR generó con éxito la región objetivo de ADN anticipada (a veces también denominada amplímero o amplicón), se puede emplear la electroforesis en gel de agarosa para la separación por tamaños de los productos de la PCR. El tamaño de los productos de la PCR se determina por comparación con una escalera de ADN, un marcador de peso molecular que contiene fragmentos de ADN de tamaños conocidos, que corre en el gel junto con los productos de la PCR.

Etapas
Al igual que con otras reacciones químicas, la velocidad de reacción y la eficiencia de la PCR se ven afectadas por factores limitantes. Por lo tanto, todo el proceso de PCR se puede dividir en tres etapas según el progreso de la reacción:
- Amplificación exponencial: en cada ciclo, la cantidad de producto se duplica (suponiendo una eficiencia de reacción del 100 %). Después de 30 ciclos, una sola copia de ADN puede incrementarse hasta 1.000.000.000 (mil millones) de copias. En cierto sentido, entonces, la replicación de una hebra discreta de ADN está siendo manipulada en un tubo bajo condiciones controladas. La reacción es muy sensible: solo deben estar presentes cantidades diminutas de ADN.
- Nivelación fuera de etapa: la reacción se ralentiza a medida que la ADN polimerasa pierde actividad y el consumo de reactivos, como dNTP y cebadores, hace que se vuelvan más limitados.
- Meseta: No se acumula más producto debido al agotamiento de los reactivos y la enzima.
Mejoramiento
En la práctica, la PCR puede fallar por varias razones, como la sensibilidad o la contaminación. La contaminación con ADN extraño puede dar lugar a productos falsos y se aborda con protocolos y procedimientos de laboratorio que separan las mezclas previas a la PCR de los posibles contaminantes del ADN. Por ejemplo, si se analiza el ADN de la escena de un crimen, una sola molécula de ADN del personal de laboratorio podría amplificarse y desviar la investigación. Por lo tanto, las áreas de configuración de PCR están separadas del análisis o la purificación de otros productos de PCR, se utiliza material de plástico desechable y la superficie de trabajo entre las configuraciones de reacción debe limpiarse a fondo.
La especificidad se puede ajustar mediante condiciones experimentales para que no se generen productos espurios. Las técnicas de diseño de cebadores son importantes para mejorar el rendimiento del producto de PCR y evitar la formación de productos inespecíficos. El uso de componentes de tampón alternativos o enzimas de polimerasa puede ayudar con la amplificación de regiones de ADN largas o problemáticas. Por ejemplo, se dice que la polimerasa Q5 es aproximadamente 280 veces menos propensa a errores que la polimerasa Taq. Tanto los parámetros de ejecución (por ejemplo, la temperatura y la duración de los ciclos) como la adición de reactivos, como la formamida, pueden aumentar la especificidad y el rendimiento de la PCR. Se pueden realizar simulaciones informáticas de los resultados teóricos de la PCR (PCR electrónica) para ayudar en el diseño del cebador.
Aplicaciones
Aislamiento selectivo de ADN
La PCR permite el aislamiento de fragmentos de ADN a partir de ADN genómico mediante la amplificación selectiva de una región específica de ADN. Este uso de PCR aumenta muchas formas, como la generación de sondas de hibridación para la hibridación del sur o del norte y la clonación de ADN, que requieren mayores cantidades de ADN, que representan una región de ADN específica. La PCR proporciona a estas técnicas grandes cantidades de ADN puro, lo que permite el análisis de muestras de ADN incluso a partir de cantidades muy pequeñas de material de partida.
Otras aplicaciones de la PCR incluyen la secuenciación de ADN para determinar secuencias amplificadas por PCR desconocidas en las que se puede usar uno de los cebadores de amplificación en la secuenciación de Sanger, el aislamiento de una secuencia de ADN para acelerar las tecnologías de ADN recombinante que involucran la inserción de una secuencia de ADN en un plásmido, fago, o cósmido (dependiendo del tamaño) o el material genético de otro organismo. Las colonias bacterianas (como E. coli) pueden examinarse rápidamente mediante PCR para detectar construcciones de vector de ADN correctas. La PCR también se puede usar para la toma de huellas genéticas; una técnica forense utilizada para identificar a una persona u organismo mediante la comparación de ADN experimental a través de diferentes métodos basados en PCR.
Algunos métodos de huellas dactilares de PCR tienen un alto poder discriminativo y pueden usarse para identificar relaciones genéticas entre individuos, como padre-hijo o entre hermanos, y se usan en pruebas de paternidad (Fig. 4). Esta técnica también se puede usar para determinar las relaciones evolutivas entre organismos cuando se usan ciertos relojes moleculares (es decir, los genes 16S rRNA y recA de microorganismos).
Amplificación y cuantificación de ADN
Debido a que la PCR amplifica las regiones de ADN a las que se dirige, la PCR se puede usar para analizar cantidades extremadamente pequeñas de muestra. Esto suele ser fundamental para el análisis forense, cuando solo se dispone de una pequeña cantidad de ADN como prueba. La PCR también se puede usar en el análisis de ADN antiguo que tiene decenas de miles de años. Estas técnicas basadas en PCR se han utilizado con éxito en animales, como un mamut de cuarenta mil años, y también en ADN humano, en aplicaciones que van desde el análisis de momias egipcias hasta la identificación de un zar ruso y el cuerpo de El rey inglés Ricardo III.
Los métodos de PCR cuantitativa o PCR en tiempo real (qPCR, que no debe confundirse con RT-PCR) permiten estimar la cantidad de una secuencia dada presente en una muestra, una técnica que a menudo se aplica para determinar cuantitativamente los niveles de expresión génica. La PCR cuantitativa es una herramienta establecida para la cuantificación de ADN que mide la acumulación de producto de ADN después de cada ronda de amplificación por PCR.
qPCR permite la cuantificación y detección de una secuencia de ADN específica en tiempo real ya que mide la concentración mientras se lleva a cabo el proceso de síntesis. Existen dos métodos para la detección y cuantificación simultáneas. El primer método consiste en utilizar colorantes fluorescentes que se retienen de forma inespecífica entre las dobles hebras. El segundo método implica sondas que codifican secuencias específicas y están marcadas con fluorescencia. La detección de ADN utilizando estos métodos solo se puede ver después de que tenga lugar la hibridación de las sondas con su ADN complementario (ADNc). Una combinación técnica interesante es la PCR en tiempo real y la transcripción inversa. Esta sofisticada técnica, denominada RT-qPCR, permite la cuantificación de una pequeña cantidad de ARN. A través de esta técnica combinada, el ARNm se convierte en ADNc, que se cuantifica aún más usando qPCR. Esta técnica reduce la posibilidad de error en el punto final de la PCR,aumentar las posibilidades de detección de genes asociados con enfermedades genéticas como el cáncer. Los laboratorios utilizan RT-qPCR con el fin de medir con sensibilidad la regulación génica. Los fundamentos matemáticos para la cuantificación confiable de PCR y RT-qPCR facilitan la implementación de procedimientos de ajuste precisos de datos experimentales en aplicaciones de investigación, médicas, diagnósticas y de enfermedades infecciosas.
Aplicaciones médicas y de diagnóstico
A los futuros padres se les puede hacer una prueba para determinar si son portadores genéticos, o se les puede hacer una prueba a sus hijos para determinar si realmente están afectados por una enfermedad. Las muestras de ADN para las pruebas prenatales se pueden obtener mediante amniocentesis, muestreo de vellosidades coriónicas o incluso mediante el análisis de células fetales raras que circulan en el torrente sanguíneo de la madre. El análisis de PCR también es esencial para el diagnóstico genético previo a la implantación, en el que se analizan las células individuales de un embrión en desarrollo para detectar mutaciones.
- La PCR también se puede utilizar como parte de una prueba sensible para la tipificación de tejidos, vital para el trasplante de órganos. A partir de 2008, incluso existe una propuesta para reemplazar las pruebas tradicionales basadas en anticuerpos para el tipo de sangre con pruebas basadas en PCR.
- Muchas formas de cáncer implican alteraciones en los oncogenes. Mediante el uso de pruebas basadas en PCR para estudiar estas mutaciones, los regímenes de terapia a veces se pueden personalizar individualmente para un paciente. La PCR permite el diagnóstico precoz de enfermedades malignas como la leucemia y los linfomas, que actualmente es la más desarrollada en la investigación del cáncer y ya se utiliza de forma rutinaria. Los ensayos de PCR se pueden realizar directamente en muestras de ADN genómico para detectar células malignas específicas de translocación con una sensibilidad que es al menos 10 000 veces mayor que la de otros métodos.La PCR es muy útil en el campo médico ya que permite el aislamiento y amplificación de supresores de tumores. La PCR cuantitativa, por ejemplo, se puede utilizar para cuantificar y analizar células individuales, así como para reconocer confirmaciones y combinaciones de ADN, ARNm y proteínas.
Aplicaciones de enfermedades infecciosas
La PCR permite un diagnóstico rápido y altamente específico de enfermedades infecciosas, incluidas las causadas por bacterias o virus. La PCR también permite la identificación de microorganismos no cultivables o de crecimiento lento, como micobacterias, bacterias anaerobias o virus a partir de ensayos de cultivo de tejidos y modelos animales. La base para las aplicaciones de diagnóstico de PCR en microbiología es la detección de agentes infecciosos y la discriminación de cepas no patógenas de patógenas en virtud de genes específicos.
La PCR ha revolucionado la caracterización y detección de organismos de enfermedades infecciosas de las siguientes maneras:
- El virus de la inmunodeficiencia humana (o VIH), es un objetivo difícil de encontrar y erradicar. Las primeras pruebas de infección se basaron en la presencia de anticuerpos contra el virus que circulan en el torrente sanguíneo. Sin embargo, los anticuerpos no aparecen hasta muchas semanas después de la infección, los anticuerpos maternos enmascaran la infección de un recién nacido y los agentes terapéuticos para combatir la infección no afectan a los anticuerpos. Se han desarrollado pruebas de PCR que pueden detectar tan solo un genoma viral entre el ADN de más de 50 000 células huésped. Las infecciones se pueden detectar antes, la sangre donada se puede analizar directamente para detectar el virus, los recién nacidos se pueden analizar inmediatamente para detectar infecciones y los efectos de los tratamientos antivirales se pueden cuantificar.
- Algunos organismos de la enfermedad, como el de la tuberculosis, son difíciles de obtener de los pacientes y su cultivo en el laboratorio es lento. Las pruebas basadas en PCR han permitido la detección de pequeñas cantidades de organismos patógenos (tanto vivos como muertos), en muestras convenientes. El análisis genético detallado también se puede utilizar para detectar la resistencia a los antibióticos, lo que permite una terapia inmediata y eficaz. Los efectos de la terapia también pueden evaluarse inmediatamente.
- La propagación de un organismo patógeno a través de poblaciones de animales domésticos o salvajes puede controlarse mediante pruebas PCR. En muchos casos, se puede detectar y monitorear la aparición de nuevos subtipos virulentos. Los subtipos de un organismo que fueron responsables de epidemias anteriores también pueden determinarse mediante análisis de PCR.
- El ADN viral puede detectarse mediante PCR. Los cebadores utilizados deben ser específicos para las secuencias objetivo en el ADN de un virus, y la PCR se puede utilizar para análisis de diagnóstico o secuenciación de ADN del genoma viral. La alta sensibilidad de la PCR permite la detección del virus poco después de la infección e incluso antes del inicio de la enfermedad. Tal detección temprana puede dar a los médicos un tiempo de anticipación significativo en el tratamiento. La cantidad de virus ("carga viral") en un paciente también se puede cuantificar mediante técnicas de cuantificación de ADN basadas en PCR (ver más abajo). Se usa una variante de PCR (RT-PCR) para detectar el ARN viral en lugar del ADN: en esta prueba, la enzima transcriptasa inversa se usa para generar una secuencia de ADN que coincide con el ARN viral; este ADN se amplifica luego según el método habitual de PCR. La RT-PCR se usa ampliamente para detectar el genoma viral del SARS-CoV-2.
- Enfermedades como la tos ferina (o tos ferina) son causadas por la bacteria Bordetella pertussis. Esta bacteria se caracteriza por una infección respiratoria aguda grave que afecta a varios animales y humanos y ha provocado la muerte de muchos niños pequeños. La toxina de la tos ferina es una exotoxina proteica que se une a los receptores celulares mediante dos dímeros y reacciona con diferentes tipos de células, como los linfocitos T, que desempeñan un papel en la inmunidad celular. La PCR es una herramienta de prueba importante que puede detectar secuencias dentro del gen de la toxina de la tos ferina. Debido a que la PCR tiene una alta sensibilidad para la toxina y un tiempo de respuesta rápido, es muy eficiente para diagnosticar la tos ferina en comparación con el cultivo.
Aplicaciones forenses
El desarrollo de protocolos de huellas dactilares genéticas (o ADN) basadas en PCR ha visto una aplicación generalizada en la medicina forense:
- En su forma más discriminatoria, la huella genética puede discriminar de manera única a cualquier persona de toda la población del mundo. Se pueden aislar muestras diminutas de ADN de la escena del crimen y compararlas con las de los sospechosos o con una base de datos de ADN de pruebas anteriores o convictos. Las versiones más simples de estas pruebas a menudo se usan para descartar rápidamente a los sospechosos durante una investigación criminal. La evidencia de crímenes de décadas de antigüedad puede ser probada, confirmando o exonerando a las personas condenadas originalmente.
- La tipificación forense de ADN ha sido una forma eficaz de identificar o exonerar a los sospechosos de delitos debido al análisis de las pruebas descubiertas en la escena del crimen. El genoma humano tiene muchas regiones repetitivas que se pueden encontrar dentro de secuencias de genes o en regiones no codificantes del genoma. Específicamente, hasta el 40% del ADN humano es repetitivo.Hay dos categorías distintas para estas regiones no codificantes repetitivas en el genoma. La primera categoría se denomina repeticiones en tándem de número variable (VNTR), que tienen una longitud de 10 a 100 pares de bases y la segunda categoría se denomina repeticiones en tándem cortas (STR) y consisten en secciones repetidas de 2 a 10 pares de bases. La PCR se usa para amplificar varios VNTR y STR bien conocidos usando cebadores que flanquean cada una de las regiones repetitivas. Los tamaños de los fragmentos obtenidos de cualquier individuo para cada uno de los STR indicarán qué alelos están presentes. Al analizar varios STR para un individuo, se encontrará un conjunto de alelos para cada persona que estadísticamente es probable que sea único.Los investigadores han identificado la secuencia completa del genoma humano. Se puede acceder fácilmente a esta secuencia a través del sitio web de NCBI y se utiliza en muchas aplicaciones de la vida real. Por ejemplo, el FBI ha compilado un conjunto de sitios de marcadores de ADN utilizados para la identificación, y estos se denominan base de datos de ADN del Sistema de índice de ADN combinado (CODIS).El uso de esta base de datos permite utilizar el análisis estadístico para determinar la probabilidad de que una muestra de ADN coincida. La PCR es una herramienta analítica muy poderosa y significativa para la tipificación forense de ADN porque los investigadores solo necesitan una cantidad muy pequeña del ADN objetivo para el análisis. Por ejemplo, un solo cabello humano con un folículo piloso adjunto tiene suficiente ADN para realizar el análisis. Del mismo modo, unos pocos espermatozoides, muestras de piel debajo de las uñas o una pequeña cantidad de sangre pueden proporcionar suficiente ADN para un análisis concluyente.
- Las formas menos discriminatorias de huellas dactilares de ADN pueden ayudar en las pruebas de paternidad de ADN, donde un individuo se compara con sus parientes cercanos. El ADN de restos humanos no identificados se puede analizar y comparar con el de posibles padres, hermanos o hijos. Se pueden usar pruebas similares para confirmar los padres biológicos de un niño adoptado (o secuestrado). El padre biológico real de un recién nacido también se puede confirmar (o descartar).
- Se ha demostrado que el diseño PCR AMGX/AMGY no solo facilita la amplificación de secuencias de ADN de una cantidad muy minúscula de genoma. Sin embargo, también se puede utilizar para la determinación del sexo en tiempo real a partir de muestras de huesos forenses. Esto proporciona una forma poderosa y efectiva de determinar el género en casos forenses y especímenes antiguos.
Aplicaciones de investigación
La PCR se ha aplicado a muchas áreas de investigación en genética molecular:
- La PCR permite la producción rápida de fragmentos cortos de ADN, incluso cuando no se conoce más que la secuencia de los dos cebadores. Esta capacidad de la PCR aumenta muchos métodos, como la generación de sondas de hibridación para la hibridación de transferencia Southern o Northern. La PCR proporciona a estas técnicas grandes cantidades de ADN puro, a veces como una sola hebra, lo que permite el análisis incluso a partir de cantidades muy pequeñas de material de partida.
- La PCR también puede ayudar a la tarea de secuenciación del ADN. Los segmentos conocidos de ADN se pueden producir fácilmente a partir de un paciente con una mutación de enfermedad genética. Las modificaciones a la técnica de amplificación pueden extraer segmentos de un genoma completamente desconocido o pueden generar solo una hebra única de un área de interés.
- PCR tiene numerosas aplicaciones para el proceso más tradicional de clonación de ADN. Puede extraer segmentos para insertarlos en un vector de un genoma más grande, que puede estar disponible solo en pequeñas cantidades. Con un único conjunto de "cebadores de vectores", también puede analizar o extraer fragmentos que ya se han insertado en vectores. Algunas alteraciones del protocolo PCR pueden generar mutaciones (generales o dirigidas al sitio) de un fragmento insertado.
- Los sitios con etiquetas de secuencia son un proceso en el que la PCR se usa como indicador de que un segmento particular de un genoma está presente en un clon particular. El Proyecto Genoma Humano encontró esta aplicación vital para mapear los clones de cósmidos que estaban secuenciando y para coordinar los resultados de diferentes laboratorios.
- Una aplicación de la PCR es el análisis filogenético del ADN de fuentes antiguas, como el que se encuentra en los huesos recuperados de los neandertales, de los tejidos congelados de los mamuts o del cerebro de las momias egipcias. En algunos casos, el ADN altamente degradado de estas fuentes podría volver a ensamblarse durante las primeras etapas de la amplificación.
- Una aplicación común de la PCR es el estudio de patrones de expresión génica. Los tejidos (o incluso las células individuales) se pueden analizar en diferentes etapas para ver qué genes se han activado o cuáles se han apagado. Esta aplicación también puede usar PCR cuantitativa para cuantificar los niveles reales de expresión
- La capacidad de la PCR para amplificar simultáneamente varios loci de espermatozoides individuales ha mejorado en gran medida la tarea más tradicional de mapeo genético mediante el estudio de cruces cromosómicos después de la meiosis. Se han observado directamente eventos de cruce raros entre loci muy cercanos mediante el análisis de miles de espermatozoides individuales. Del mismo modo, se pueden analizar deleciones, inserciones, translocaciones o inversiones inusuales, todo ello sin tener que esperar (o pagar) los largos y laboriosos procesos de fecundación, embriogénesis, etc.
- Mutagénesis dirigida al sitio: la PCR se puede usar para crear genes mutantes con mutaciones elegidas por los científicos a voluntad. Estas mutaciones se pueden elegir para comprender cómo las proteínas cumplen sus funciones y para cambiar o mejorar la función de la proteína.
Ventajas
PCR tiene una serie de ventajas. Es bastante simple de entender y usar, y produce resultados rápidamente. La técnica es muy sensible y tiene el potencial de producir de millones a miles de millones de copias de un producto específico para secuenciación, clonación y análisis. qRT-PCR comparte las mismas ventajas que la PCR, con la ventaja añadida de la cuantificación del producto sintetizado. Por lo tanto, tiene sus usos para analizar alteraciones de los niveles de expresión génica en tumores, microbios u otros estados patológicos.
PCR es una herramienta de investigación muy poderosa y práctica. La PCR está determinando la secuenciación de etiologías desconocidas de muchas enfermedades. La técnica puede ayudar a identificar la secuencia de virus previamente desconocidos relacionados con los ya conocidos y así darnos una mejor comprensión de la enfermedad en sí. Si el procedimiento se puede simplificar aún más y se pueden desarrollar sistemas sensibles de detección no radiométrica, la PCR ocupará un lugar destacado en el laboratorio clínico en los años venideros.
Limitaciones
Una de las principales limitaciones de la PCR es que se necesita información previa sobre la secuencia diana para generar los cebadores que permitirán su amplificación selectiva. Esto significa que, por lo general, los usuarios de PCR deben conocer la(s) secuencia(s) precisa(s) aguas arriba de la región objetivo en cada una de las dos plantillas monocatenarias para garantizar que la ADN polimerasa se una correctamente a los híbridos cebador-plantilla y, posteriormente, genere la toda la región diana durante la síntesis de ADN.
Como todas las enzimas, las ADN polimerasas también son propensas a errores, lo que a su vez provoca mutaciones en los fragmentos de PCR que se generan.
Otra limitación de la PCR es que se puede amplificar incluso la cantidad más pequeña de ADN contaminante, lo que genera resultados engañosos o ambiguos. Para minimizar la posibilidad de contaminación, los investigadores deben reservar salas separadas para la preparación de reactivos, la PCR y el análisis del producto. Los reactivos deben dispensarse en alícuotas de un solo uso. Deben utilizarse de forma rutinaria pipetas con émbolos desechables y puntas de pipeta extralargas. Además, se recomienda asegurarse de que la configuración del laboratorio siga un flujo de trabajo unidireccional. Ningún material o reactivo utilizado en las salas de análisis y PCR debe llevarse nunca a la sala de preparación de PCR sin una descontaminación completa.
Las muestras ambientales que contienen ácidos húmicos pueden inhibir la amplificación por PCR y generar resultados inexactos.
Variaciones
- PCR específica de alelo: una técnica de diagnóstico o clonación basada en variaciones de un solo nucleótido (SNV que no deben confundirse con SNP) (diferencias de una sola base en un paciente). Requiere un conocimiento previo de una secuencia de ADN, incluidas las diferencias entre los alelos, y utiliza cebadores cuyos extremos 3' engloban el SNV (par de bases amortiguadoras alrededor del SNV normalmente incorporado). La amplificación por PCR en condiciones estrictas es mucho menos eficiente en presencia de un desajuste entre la plantilla y el cebador, por lo que la amplificación exitosa con un cebador específico de SNP señala la presencia del SNP específico en una secuencia. Consulte Genotipado de SNP para obtener más información.
- PCR de ensamblaje o ensamblaje cíclico de polimerasa (PCA): síntesis artificial de secuencias de ADN largas mediante la realización de PCR en un conjunto de oligonucleótidos largos con segmentos superpuestos cortos. Los oligonucleótidos alternan entre direcciones con sentido y antisentido, y los segmentos superpuestos determinan el orden de los fragmentos de PCR, produciendo así selectivamente el producto de ADN largo final.
- PCR asimétrica: amplifica preferentemente una cadena de ADN en una plantilla de ADN de doble cadena. Se utiliza en la secuenciación y el sondeo de hibridación donde se requiere la amplificación de solo una de las dos cadenas complementarias. La PCR se lleva a cabo como de costumbre, pero con un gran exceso del cebador para la cadena objetivo de la amplificación. Debido a la lenta amplificación (aritmética) posterior en la reacción después de que se agotó el cebador limitante, se requieren ciclos adicionales de PCR. Una modificación reciente de este proceso, conocida como L inear - A fter - The- E xponential -PCR (LATE-PCR), utiliza un cebador limitante con una temperatura de fusión más alta (T m) que el cebador en exceso para mantener la eficiencia de la reacción a medida que la concentración del cebador limitante disminuye a mitad de la reacción.
- PCR convectiva: una forma pseudo-isotérmica de realizar la PCR. En lugar de calentar y enfriar repetidamente la mezcla de PCR, la solución se somete a un gradiente térmico. El flujo convectivo resultante impulsado por la inestabilidad térmica mezcla automáticamente los reactivos de PCR de las regiones frías y calientes, lo que permite la PCR repetidamente. Los parámetros como las condiciones de contorno térmico y la geometría del recinto de la PCR se pueden optimizar para producir una PCR robusta y rápida aprovechando la aparición de campos de flujo caóticos. Tal configuración de PCR de flujo convectivo reduce significativamente el requisito de energía del dispositivo y el tiempo de operación.
- Dial-out PCR: un método altamente paralelo para recuperar moléculas de ADN precisas para la síntesis de genes. Una biblioteca compleja de moléculas de ADN se modifica con etiquetas flanqueantes únicas antes de la secuenciación paralela masiva. Los cebadores dirigidos por etiquetas permiten la recuperación de moléculas con las secuencias deseadas mediante PCR.
- PCR digital (dPCR): se utiliza para medir la cantidad de una secuencia de ADN diana en una muestra de ADN. La muestra de ADN está muy diluida, de modo que después de ejecutar muchas PCR en paralelo, algunas de ellas no reciben ni una sola molécula del ADN objetivo. La concentración de ADN objetivo se calcula utilizando la proporción de resultados negativos. De ahí el nombre de 'PCR digital'.
- Amplificación dependiente de helicasa: similar a la PCR tradicional, pero utiliza una temperatura constante en lugar de ciclos de desnaturalización y recocido/extensión. La ADN helicasa, una enzima que desenrolla el ADN, se utiliza en lugar de la desnaturalización térmica.
- PCR de inicio en caliente: una técnica que reduce la amplificación no específica durante las etapas iniciales de configuración de la PCR. Puede realizarse manualmente calentando los componentes de la reacción a la temperatura de desnaturalización (p. ej., 95 °C) antes de añadir la polimerasa. Se han desarrollado sistemas enzimáticos especializados que inhiben la actividad de la polimerasa a temperatura ambiente, ya sea por la unión de un anticuerpo o por la presencia de inhibidores unidos covalentemente que se disocian solo después de un paso de activación a alta temperatura. La PCR de inicio en caliente/final en frío se logra con nuevas polimerasas híbridas que son inactivas a temperatura ambiente y se activan instantáneamente a la temperatura de elongación.
- La PCR in silico (PCR digital, PCR virtual, PCR electrónica, e-PCR) se refiere a las herramientas informáticas utilizadas para calcular los resultados teóricos de la reacción en cadena de la polimerasa utilizando un conjunto determinado de cebadores (sondas) para amplificar las secuencias de ADN de un genoma secuenciado o transcriptoma. La PCR in silico se propuso como una herramienta educativa para la biología molecular.
- PCR específica entre secuencias (ISSR): un método de PCR para la toma de huellas dactilares de ADN que amplifica regiones entre repeticiones de secuencias simples para producir una huella digital única de longitudes de fragmentos amplificados.
- PCR inversa: se usa comúnmente para identificar las secuencias flanqueantes alrededor de los insertos genómicos. Implica una serie de digestiones de ADN y autoligado, lo que da como resultado secuencias conocidas en cada extremo de la secuencia desconocida.
- PCR mediada por ligación: utiliza pequeños conectores de ADN ligados al ADN de interés y múltiples cebadores que se unen a los conectores de ADN; se ha utilizado para la secuenciación de ADN, la caminata genómica y la huella de ADN.
- PCR específica de metilación (MSP): desarrollada por Stephen Baylin y James G. Herman en la Escuela de Medicina Johns Hopkins, y se usa para detectar la metilación de las islas CpG en el ADN genómico. El ADN se trata primero con bisulfito de sodio, que convierte las bases de citosina no metiladas en uracilo, que los cebadores de PCR reconocen como timina. Luego se llevan a cabo dos PCR en el ADN modificado, usando conjuntos de cebadores idénticos excepto en cualquier isla CpG dentro de las secuencias de cebadores. En estos puntos, un conjunto de cebadores reconoce el ADN con citosinas para amplificar el ADN metilado y un conjunto reconoce el ADN con uracilo o timina para amplificar el ADN no metilado. MSP usando qPCR también se puede realizar para obtener información cuantitativa en lugar de cualitativa sobre la metilación.
- Miniprimer PCR: utiliza una polimerasa termoestable (S-Tbr) que puede extenderse desde cebadores cortos ("smalligos") tan cortos como 9 o 10 nucleótidos. Este método permite que la PCR se dirija a regiones de unión de cebadores más pequeñas y se usa para amplificar secuencias de ADN conservadas, como el gen de ARNr 16S (o eucariota 18S).
- Amplificación de sonda dependiente de ligación multiplex (MLPA): permite amplificar múltiples objetivos con un solo par de cebadores, evitando así las limitaciones de resolución de la PCR multiplex (ver más abajo).
- Multiplex-PCR: consta de múltiples conjuntos de cebadores dentro de una sola mezcla de PCR para producir amplicones de diferentes tamaños que son específicos para diferentes secuencias de ADN. Al apuntar a múltiples genes a la vez, se puede obtener información adicional de una sola prueba que, de otro modo, requeriría varios reactivos y más tiempo para realizarse. Las temperaturas de recocido para cada uno de los conjuntos de cebadores deben optimizarse para que funcionen correctamente dentro de una sola reacción y los tamaños de amplicón. Es decir, la longitud de sus pares de bases debe ser lo suficientemente diferente para formar bandas distintas cuando se visualice mediante electroforesis en gel.
- PCR asistida por nanopartículas (nanoPCR): algunas nanopartículas (NP) pueden mejorar la eficiencia de la PCR (por lo que se denominan nanoPCR), y algunas incluso pueden superar a los potenciadores de PCR originales. Se informó que los puntos cuánticos (QD) pueden mejorar la especificidad y la eficiencia de la PCR. Los nanotubos de carbono de pared simple (SWCNT) y los nanotubos de carbono de pared múltiple (MWCNT) son eficaces para mejorar la amplificación de la PCR larga. El nanopolvo de carbono (CNP) puede mejorar la eficiencia de la PCR repetida y la PCR larga, mientras que se descubrió que el óxido de zinc, el dióxido de titanio y las NP de Ag aumentan el rendimiento de la PCR. Los datos anteriores indicaron que las NP no metálicas conservaron una fidelidad de amplificación aceptable. Dado que muchas NP son capaces de mejorar la eficiencia de la PCR, está claro que es probable que exista un gran potencial para las mejoras tecnológicas y el desarrollo de productos de nanoPCR.
- PCR anidada: aumenta la especificidad de la amplificación del ADN al reducir el fondo debido a la amplificación no específica del ADN. Se utilizan dos conjuntos de cebadores en dos PCR sucesivas. En la primera reacción, se usa un par de cebadores para generar productos de ADN que, además del objetivo previsto, aún pueden consistir en fragmentos de ADN amplificados no específicamente. A continuación, los productos se utilizan en una segunda PCR con un conjunto de cebadores cuyos sitios de unión son total o parcialmente diferentes y están situados en 3' de cada uno de los cebadores utilizados en la primera reacción. La PCR anidada suele tener más éxito en la amplificación específica de fragmentos largos de ADN que la PCR convencional, pero requiere un conocimiento más detallado de las secuencias objetivo.
- PCR de extensión superpuesta o empalme por extensión superpuesta (SOEing): una técnica de ingeniería genética que se utiliza para empalmar dos o más fragmentos de ADN que contienen secuencias complementarias. Se utiliza para unir piezas de ADN que contienen genes, secuencias reguladoras o mutaciones; la técnica permite la creación de construcciones de ADN largas y específicas. También puede introducir deleciones, inserciones o mutaciones puntuales en una secuencia de ADN.
- PAN-AC: utiliza condiciones isotérmicas para la amplificación y puede utilizarse en células vivas.
- PCR cuantitativa (qPCR): se utiliza para medir la cantidad de una secuencia objetivo (comúnmente en tiempo real). Mide cuantitativamente las cantidades iniciales de ADN, ADNc o ARN. La PCR cuantitativa se usa comúnmente para determinar si una secuencia de ADN está presente en una muestra y el número de sus copias en la muestra. La PCR cuantitativa tiene un grado de precisión muy alto. Los métodos de PCR cuantitativa utilizan tintes fluorescentes, como Sybr Green, EvaGreen o sondas de ADN que contienen fluoróforos, como TaqMan, para medir la cantidad de producto amplificado en tiempo real. A veces también se abrevia como RT-PCR (PCR en tiempo real), pero esta abreviatura debe usarse solo para PCR de transcripción inversa. qPCR son las contracciones apropiadas para la PCR cuantitativa (PCR en tiempo real).
- PCR de complemento inverso (RC-PCR): permite añadir dominios funcionales o secuencias de elección de forma independiente a cualquiera de los extremos del amplicón generado en una sola reacción de tubo cerrado. Este método genera cebadores específicos de diana dentro de la reacción mediante la interacción de cebadores universales (que contienen las secuencias o dominios deseados que se agregarán) y sondas RC.
- PCR de transcripción inversa (RT-PCR): para amplificar ADN a partir de ARN. La transcriptasa inversa transcribe inversamente el ARN en ADNc, que luego se amplifica mediante PCR. La RT-PCR se usa ampliamente en el perfilado de expresión, para determinar la expresión de un gen o para identificar la secuencia de una transcripción de ARN, incluidos los sitios de inicio y terminación de la transcripción. Si se conoce la secuencia de ADN genómico de un gen, se puede usar RT-PCR para mapear la ubicación de exones e intrones en el gen. El extremo 5' de un gen (correspondiente al sitio de inicio de la transcripción) se identifica normalmente mediante RACE-PCR (amplificación rápida de extremos de cDNA).
- PCR dependiente de RNasa H (rhPCR): una modificación de la PCR que utiliza cebadores con un bloque de extensión 3' que puede eliminarse mediante una enzima RNasa HII termoestable. Este sistema reduce los dímeros de cebadores y permite realizar reacciones multiplexadas con un mayor número de cebadores.
- Single Specific Primer-PCR (SSP-PCR): permite la amplificación de ADN de doble cadena incluso cuando la información de la secuencia está disponible en un solo extremo. Este método permite la amplificación de genes para los que solo se dispone de información de secuencia parcial, y permite que el genoma pase unidireccionalmente de regiones conocidas a regiones desconocidas del cromosoma.
- PCR en fase sólida: abarca múltiples significados, incluida la amplificación de polonia (donde las colonias de PCR se derivan en una matriz de gel, por ejemplo), PCR puente (los cebadores se unen covalentemente a una superficie de soporte sólido), PCR en fase sólida convencional (donde la PCR asimétrica es aplicado en presencia de un cebador que contiene un soporte sólido con una secuencia que coincide con uno de los cebadores acuosos) y PCR en fase sólida mejorada (donde la PCR en fase sólida convencional se puede mejorar mediante el empleo de un cebador de soporte sólido anidado y Tm alta con la aplicación opcional de un "paso" térmico para favorecer la imprimación de soportes sólidos).
- Suicide PCR: normalmente se utiliza en paleogenética u otros estudios en los que evitar falsos positivos y garantizar la especificidad del fragmento amplificado es la máxima prioridad. Se describió originalmente en un estudio para verificar la presencia del microbio Yersinia pestis en muestras dentales obtenidas de tumbas del siglo XIV de personas supuestamente muertas por la peste durante la epidemia medieval de Peste Negra.El método prescribe el uso de cualquier combinación de cebadores solo una vez en una PCR (de ahí el término "suicidio"), que nunca debería haberse usado en ninguna reacción de PCR de control positivo, y los cebadores siempre deben apuntar a una región genómica nunca amplificada antes en el laboratorio utilizando este o cualquier otro conjunto de cebadores. Esto garantiza que no haya ADN contaminante de reacciones de PCR anteriores en el laboratorio, que de lo contrario podría generar falsos positivos.
- PCR térmica asimétrica entrelazada (TAIL-PCR): para el aislamiento de una secuencia desconocida que flanquea una secuencia conocida. Dentro de la secuencia conocida, TAIL-PCR utiliza un par de cebadores anidados con diferentes temperaturas de hibridación; se usa un cebador degenerado para amplificar en la otra dirección desde la secuencia desconocida.
- Touchdown PCR (Step-down PCR): una variante de PCR que tiene como objetivo reducir el fondo no específico al reducir gradualmente la temperatura de hibridación a medida que avanza el ciclo de PCR. La temperatura de recocido en los ciclos iniciales suele estar unos pocos grados (3–5 °C) por encima de la T m de los cebadores utilizados, mientras que en los ciclos posteriores está unos pocos grados (3–5 °C) por debajo de la T del cebador. m _ Las temperaturas más altas dan una mayor especificidad para la unión del cebador y las temperaturas más bajas permiten una amplificación más eficiente de los productos específicos formados durante los ciclos iniciales.
- Universal Fast Walking: para la caminata genómica y la toma de huellas dactilares genéticas utilizando una PCR 'bilateral' más específica que los enfoques 'unilaterales' convencionales (utilizando solo un cebador específico del gen y un cebador general, lo que puede conducir a un 'ruido' artificial) en virtud de un mecanismo que implica la formación de estructuras de lariat. Los derivados optimizados de UFW son LaNe RAGE (PCR anidada dependiente de lariat para la amplificación rápida de extremos de ADN genómico), 5'RACE LaNe y 3'RACE LaNe.
Historia
Las enzimas resistentes al calor que son un componente clave en la reacción en cadena de la polimerasa se descubrieron en la década de 1960 como producto de una forma de vida microbiana que vivía en las aguas sobrecalentadas del Mushroom Spring de Yellowstone.
Un artículo de 1971 en el Journal of Molecular Biology de Kjell Kleppe y colaboradores en el laboratorio de H. Gobind Khorana describió por primera vez un método de uso de un ensayo enzimático para replicar una plantilla corta de ADN con cebadores in vitro. Sin embargo, esta primera manifestación del principio básico de PCR no recibió mucha atención en ese momento y la invención de la reacción en cadena de la polimerasa en 1983 generalmente se atribuye a Kary Mullis.
Cuando Mullis desarrolló la PCR en 1983, trabajaba en Emeryville, California para Cetus Corporation, una de las primeras empresas de biotecnología, donde era responsable de sintetizar cadenas cortas de ADN. Mullis ha escrito que concibió la idea de PCR mientras recorría la Pacific Coast Highway una noche en su automóvil. Estaba jugando en su mente con una nueva forma de analizar los cambios (mutaciones) en el ADN cuando se dio cuenta de que, en cambio, había inventado un método para amplificar cualquier región del ADN a través de ciclos repetidos de duplicación impulsados por la ADN polimerasa. en cientifico americano, Mullis resumió el procedimiento: "Comenzando con una sola molécula del material genético ADN, la PCR puede generar 100 mil millones de moléculas similares en una tarde. La reacción es fácil de ejecutar. No requiere más que un tubo de ensayo, algunos reactivos simples y una fuente de calor". La toma de huellas dactilares de ADN se utilizó por primera vez para las pruebas de paternidad en 1988.
Mullis ha acreditado su uso de LSD como parte integral de su desarrollo de la PCR: "¿Habría inventado la PCR si no hubiera tomado LSD? Lo dudo seriamente. Podría sentarme en una molécula de ADN y ver pasar los polímeros. Aprendí eso en parte con drogas psicodélicas".
Mullis y el bioquímico Michael Smith, que había desarrollado otras formas esenciales de manipular el ADN, recibieron conjuntamente el Premio Nobel de Química en 1993, siete años después de que Mullis y sus colegas de Cetus pusieran en práctica su propuesta por primera vez. El artículo de Mullis de 1985 con RK Saiki y HA Erlich, "Enzymatic Amplification of β-globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia", la invención de la reacción en cadena de la polimerasa (PCR), fue honrado con una Citación por el premio Chemical Breakthrough Award de la División de Historia de la Química de la American Chemical Society en 2017.
El núcleo del método de PCR es el uso de una ADN polimerasa adecuada capaz de soportar las altas temperaturas de >90 °C (194 °F) requeridas para la separación de las dos cadenas de ADN en la doble hélice de ADN después de cada ciclo de replicación. Las polimerasas de ADN empleadas inicialmente para experimentos in vitro que presagiaban PCR no pudieron soportar estas altas temperaturas. Por lo tanto, los primeros procedimientos para la replicación del ADN eran muy ineficientes y requerían mucho tiempo, y requerían grandes cantidades de ADN polimerasa y un manejo continuo durante todo el proceso.
El descubrimiento en 1976 de la Taq polimerasa, una ADN polimerasa purificada de la bacteria termófila Thermus aquaticus, que vive naturalmente en ambientes cálidos (50 a 80 °C (122 a 176 °F)) como las fuentes termales, allanó el camino para mejoras del método PCR. La ADN polimerasa aislada de T. aquaticus es estable a altas temperaturas y permanece activa incluso después de la desnaturalización del ADN, lo que evita la necesidad de agregar nueva ADN polimerasa después de cada ciclo. Esto permitió un proceso basado en un termociclador automatizado para la amplificación de ADN.
Disputas de patentes
La técnica de PCR fue patentada por Kary Mullis y cedida a Cetus Corporation, donde trabajaba Mullis cuando inventó la técnica en 1983. La enzima polimerasa Taq también estaba cubierta por patentes. Ha habido varias demandas de alto perfil relacionadas con la técnica, incluida una demanda sin éxito presentada por DuPont. La compañía farmacéutica suiza Hoffmann-La Roche compró los derechos de las patentes en 1992. La última de las patentes comerciales de PCR expiró en 2017.
Una batalla relacionada con la patente sobre la enzima polimerasa Taq todavía está en curso en varias jurisdicciones de todo el mundo entre Roche y Promega. Los argumentos legales se han extendido más allá de las vidas de las patentes originales de polimerasa Taq y PCR, que expiraron el 28 de marzo de 2005.
Contenido relacionado
Gas pobre
Sistema de confianza
Ferrocarril de vía ancha