
Sustitución aromática electrófila
La sustitución aromática electrófila es una reacción orgánica en la que un átomo que está unido a un sistema aromático (generalmente hidrógeno) es reemplazado por un electrófilo. Algunas de las sustituciones aromáticas electrofílicas más importantes son la nitración aromática, la halogenación aromática, la sulfonación aromática y la reacción de Friedel-Crafts de alquilación y acilación.
Reacciones ilustrativas
El ejemplo más practicado de esta reacción es la etilación del benceno.

En 1999 se produjeron aproximadamente 24.700.000 toneladas. (Después de la deshidrogenación y la polimerización, se produce el poliestireno plástico básico). En este proceso, se utilizan ácidos como catalizadores para generar el carbocatión incipiente. Se llevan a cabo muchas otras reacciones electrofílicas del benceno, aunque en una escala mucho menor; son rutas valiosas hacia intermediarios clave. La nitración del benceno se logra mediante la acción del ion nitronio como electrófilo. La sulfonación con ácido sulfúrico fumante da ácido bencenosulfónico. La halogenación aromática con bromo, cloro o yodo da los haluros de arilo correspondientes. Esta reacción normalmente es catalizada por el trihaluro de hierro o aluminio correspondiente.

La reacción de Friedel-Crafts se puede realizar como acilación o como alquilación. A menudo, se usa tricloruro de aluminio, pero se puede aplicar casi cualquier ácido de Lewis fuerte. Para la reacción de acilación se requiere una cantidad estequiométrica de tricloruro de aluminio.
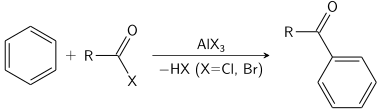

Mecanismo de reacción
El mecanismo de reacción general, denotado por el símbolo mecanicista de Hughes-Ingold S E Ar, comienza cuando el anillo aromático ataca al electrófilo E. Este paso conduce a la formación de un catión ciclohexadienilo cargado positivamente y deslocalizado, también conocido como ion arenio, Wheland intermedio o areno σ-complejo. Se han caracterizado muchos ejemplos de este carbocatión, pero en condiciones normales de operación, estas especies altamente ácidas donarán el protón unido al carbono sp al solvente (o cualquier otra base débil) para restablecer la aromaticidad. El resultado neto es el reemplazo de H por E en el anillo de arilo. Ocasionalmente, otros electrófugos (grupos que pueden salir sin su par de electrones) al lado de Hpartirá para restablecer la aromaticidad; estas especies incluyen grupos sililo (como SiR 3), el grupo carboxi (como CO 2 + H), el grupo yodo (como I) y grupos alquilo terciario como t -butilo (como R). La capacidad de este tipo de sustituyentes para salir es a veces explotada sintéticamente, particularmente en el caso de reemplazo de sililo por otro grupo funcional (ataque ipso). Sin embargo, la pérdida de grupos como yodo o alquilo suele ser una reacción secundaria no deseada.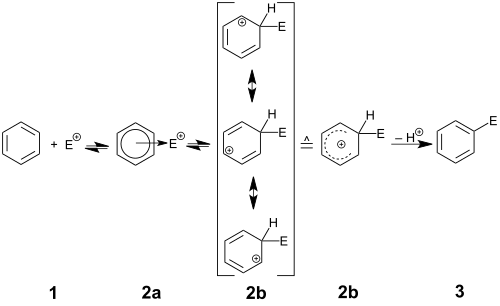
Efecto de los grupos sustituyentes
Tanto la regioselectividad (los diversos patrones de sustitución de areno) como la velocidad de una sustitución aromática electrófila se ven afectadas por los sustituyentes ya unidos al anillo de benceno. En términos de regioselectividad, algunos grupos promueven la sustitución en las posiciones orto o para, mientras que otros grupos favorecen la sustitución en la posición meta. Estos grupos se denominan dirección orto-para o dirección meta, respectivamente. Además, algunos grupos aumentarán la velocidad de reacción (activando) mientras que otros disminuirán la velocidad (desactivando). Mientras que los patrones de regioselectividad pueden explicarse con estructuras de resonancia, la influencia en la cinética puede explicarse tanto por estructuras de resonancia como por el efecto inductivo.
Tasa de reacción
Los sustituyentes generalmente se pueden dividir en dos clases con respecto a la sustitución electrofílica: activación y desactivación hacia el anillo aromático. Los sustituyentes activadores o los grupos activadores estabilizan el intermedio catiónico formado durante la sustitución mediante la donación de electrones al sistema de anillos, ya sea por efecto inductivo o efectos de resonancia. Ejemplos de anillos aromáticos activados son tolueno, anilina y fenol.
La densidad de electrones adicional entregada al anillo por el sustituyente no se distribuye uniformemente por todo el anillo, sino que se concentra en los átomos 2, 4 y 6, por lo que los sustituyentes activadores también son directores orto/para (ver más abajo).
Por otro lado, los sustituyentes desactivadoresdesestabilizar el catión intermedio y así disminuir la velocidad de reacción por efectos inductivos o de resonancia. Lo hacen retirando la densidad de electrones del anillo aromático. La desactivación del sistema aromático significa que, en general, se requieren condiciones más duras para que la reacción se complete. Un ejemplo de esto es la nitración de tolueno durante la producción de trinitrotolueno (TNT). Mientras que la primera nitración, sobre el anillo de tolueno activado, se puede hacer a temperatura ambiente y con ácido diluido, la segunda, sobre el anillo de nitrotolueno desactivado, ya necesita un calentamiento prolongado y ácido más concentrado, y la tercera, sobre un anillo de nitrotolueno desactivado muy fuertemente. dinitrotolueno, tiene que hacerse en ácido sulfúrico concentrado hirviendo. Los grupos que atraen electrones por resonancia disminuyen la densidad de electrones, especialmente en las posiciones 2, 4 y 6, dejando las posiciones 3 y 5 como las de reactividad comparativamente más alta, por lo que este tipo de grupos son metadirectores (ver más abajo). Los halógenos son electronegativos, por lo que se desactivan por inducción, pero tienen pares solitarios, por lo que son donantes de resonancia y, por lo tanto, directores orto/para.
Directores de orto/para
Los grupos con pares de electrones no compartidos, como el grupo amino de la anilina, se activan fuertemente y son de dirección orto/para por resonancia. Dichos grupos activadores donan esos electrones no compartidos al sistema pi, creando una carga negativa en las posiciones orto y para. Estas posiciones son, por lo tanto, las más reactivas hacia un electrófilo pobre en electrones. Esta mayor reactividad podría compensarse con el impedimento estérico entre el grupo activador y el electrófilo, pero por otro lado hay dos posiciones orto para la reacción pero solo una posición para. Por lo tanto, el resultado final de la sustitución aromática electrófila es difícil de predecir y, por lo general, solo se establece haciendo la reacción y observando la relación de sustitución orto versus para.

Además de la naturaleza nucleófila aumentada del anillo original, cuando el electrófilo ataca las posiciones orto y para de la anilina, el átomo de nitrógeno puede donar densidad de electrones al sistema pi (formando un ion iminio), dando cuatro estructuras de resonancia (a diferencia de tres en la reacción básica). Esto mejora sustancialmente la estabilidad del intermedio catiónico.

Cuando el electrófilo ataca la posición meta, el átomo de nitrógeno no puede donar densidad de electrones al sistema pi, dando solo tres contribuyentes de resonancia. Este razonamiento es consistente con los bajos rendimientos del producto sustituido en meta.

Otros sustituyentes, como los sustituyentes alquilo y arilo, también pueden donar densidad electrónica al sistema pi; sin embargo, dado que carecen de un par de electrones no compartidos disponibles, su capacidad para hacer esto es bastante limitada. Por lo tanto, solo activan débilmente el anillo y no desfavorecen fuertemente la posición meta.
La ortometalación dirigida es un tipo especial de EAS con directores de orto especiales.
Metadirectores _
Los grupos no halógenos con átomos que son más electronegativos que el carbono, como un grupo de ácido carboxílico (-CO 2 H), retiran una densidad de electrones sustancial del sistema pi. Estos grupos son grupos fuertemente desactivadores. Además, dado que el carbono sustituido ya es pobre en electrones, cualquier estructura que tenga un contribuyente de resonancia en el que haya una carga positiva en el carbono que lleva el grupo atractor de electrones (es decir, ataque orto o para) es menos estable que las otras. Por lo tanto, estos grupos atractores de electrones son metadirigidos porque esta es la posición que no tiene tanta desestabilización.
La reacción también es mucho más lenta (una velocidad de reacción relativa de 6 × 10 en comparación con el benceno) porque el anillo es menos nucleofílico.
Reacción sobre la piridina
En comparación con el benceno, la tasa de sustitución electrofílica de la piridina es mucho más lenta debido a la mayor electronegatividad del átomo de nitrógeno. Además, el nitrógeno en la piridina adquiere fácilmente una carga positiva ya sea por protonación (por nitración o sulfonación) o por ácidos de Lewis (como AlCl 3) que se usan para catalizar la reacción. Esto hace que la reacción sea aún más lenta al tener cargas formales adyacentes en el carbono y el nitrógeno o 2 cargas formales en un átomo localizado. Hacer una sustitución electrofílica directamente en piridina es casi imposible.
Para hacer la reacción se pueden hacer por 2 reacciones posibles, ambas indirectas.
Una forma posible de hacer una sustitución de piridina es la sustitución aromática nucleófila. Incluso sin catalizadores, el átomo de nitrógeno, al ser electronegativo, puede mantener la carga negativa por sí mismo. Otra forma es hacer una oxidación antes de la sustitución electrofílica. Esto produce N -óxido de piridina, que debido al átomo de oxígeno negativo, hace que la reacción sea más rápida que la piridina e incluso el benceno. Luego, el óxido se puede reducir a la piridina sustituida.
ataque ipso
La unión de un grupo entrante a una posición en un compuesto aromático que ya lleva un grupo sustituyente (que no sea hidrógeno). El grupo entrante puede desplazar a ese grupo sustituyente pero también puede ser expulsado o migrar a otra posición en un paso posterior. No se utiliza el término ' ipso -sustitución', ya que es sinónimo de sustitución. Un ejemplo clásico es la reacción del ácido salicílico con una mezcla de ácido nítrico y sulfúrico para formar ácido pícrico. La nitración de la posición 2 implica la pérdida de CO 2 como grupo saliente. La desulfonación en la que un grupo sulfonilo se sustituye por un protón es un ejemplo común. Véase también reordenamiento de Hayashi. En aromáticos sustituidos por silicio, el silicio reacciona por sustitución ipso.
Heterociclos de cinco miembros
En comparación con el benceno, los furanos, los tiofenos y los pirroles son más susceptibles al ataque electrofílico. Todos estos compuestos contienen un átomo con un par de electrones no compartido (oxígeno, azufre o nitrógeno) como miembro del anillo aromático, que estabiliza sustancialmente el intermedio catiónico. Ejemplos de sustituciones electrófilas del pirrol son la reacción de Pictet-Spengler y la reacción de Bischler-Napieralski.
Sustitución aromática electrófila asimétrica
Las sustituciones aromáticas electrófilas con electrófilos de carbono proquirales se han adaptado para la síntesis asimétrica cambiando a catalizadores ácidos de Lewis quirales, especialmente en reacciones de tipo Friedel-Crafts. Un ejemplo temprano se refiere a la adición de cloral a fenoles catalizada por cloruro de aluminio modificado con (–)-mentol. Se ha agregado un compuesto de glioxilato a la N, N-dimetilanilina con un sistema de catalizador de triflato de cobre (II) ligando de bisoxazolina quiral también en una hidroxialquilación de Friedel-Crafts:

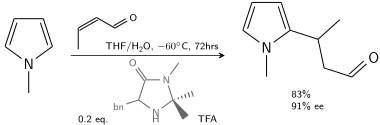
En otra alquilación, el N-metilpirrol reacciona con crotonaldehído catalizado por ácido trifluoroacético modificado con una imidazolidinona quiral:
El indol reacciona con una enamida catalizada por un ácido fosfórico derivado de BINOL quiral:
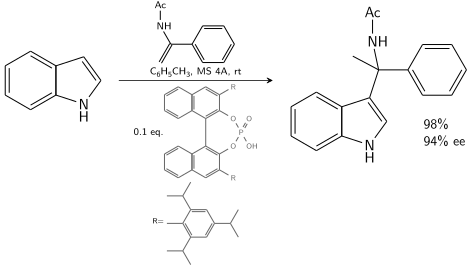
En presencia de un 10–20 % de catalizador quiral, se puede lograr un 80–90 % de ee.
Otras reacciones
- Otras reacciones que siguen un patrón de sustitución aromática electrofílica son un grupo de reacciones de formilación aromática que incluyen la reacción de Vilsmeier-Haack, la reacción de Gattermann Koch y la reacción de Reimer-Tiemann.
- Otros electrófilos son sales aromáticas de diazonio en acoplamientos de diazonio, dióxido de carbono en la reacción de Kolbe-Schmitt y grupos carbonilo activados en la condensación de Pechmann, iones de hidroxicarbenio en la clorometilación de Blanc a través de un (hidroximetil) areno intermedio (alcohol bencílico), catión clorilo (ClO 3) para la perclorilación electrofílica.
- En la reacción de varios pasos de Lehmstedt-Tanasescu, uno de los electrófilos es un intermedio N-nitroso.
- En la reacción de Tscherniac-Einhorn (llamada así por Joseph Tscherniac y Alfred Einhorn), el electrófilo es un derivado de N-metanol de una amida.
Contenido relacionado
Péptido de cobre GHK-Cu
Silicato
Estructura química