
SMILES
El sistema de entrada de línea de entrada molecular simplificado (SMILES, por simplified molecular-input line-entry system) es una especificación en forma de notación de línea para describir la estructura de especies químicas utilizando cadenas ASCII cortas. La mayoría de los editores de moléculas pueden importar cadenas SMILES para volver a convertirlas en dibujos bidimensionales o modelos tridimensionales de las moléculas.
La especificación original de SONRISAS se inició en la década de 1980. Desde entonces ha sido modificado y ampliado. En 2007, se desarrolló un estándar abierto llamado OpenSMILES en la comunidad química de código abierto.
Historia
La especificación SMILES original fue iniciada por David Weininger en el Laboratorio de la División de Ecología del Medio Continente de la USEPA en Duluth en la década de 1980. Fueron reconocidos por su participación en el desarrollo inicial "Gilman Veith y Rose Russo (USEPA) y Albert Leo y Corwin Hansch (Pomona College) por apoyar el trabajo, y Arthur Weininger (Pomona; Daylight CIS) y Jeremy Scofield (Cedar River Software, Renton, WA) por su asistencia en la programación del sistema". La Agencia de Protección Ambiental financió el proyecto inicial para desarrollar SONRISAS.
Desde entonces, ha sido modificado y ampliado por otros, sobre todo por Daylight Chemical Information Systems. En 2007, la comunidad química de código abierto Blue Obelisk desarrolló un estándar abierto llamado "OpenSMILES". Otras notaciones 'lineales' incluyen Wiswesser Line Notation (WLN), ROSDAL y SLN (Tripos Inc).
En julio de 2006, la IUPAC introdujo el InChI como estándar para la representación de fórmulas. Generalmente se considera que SMILES tiene la ventaja de ser más legible por humanos que InChI; también tiene una amplia base de soporte de software con amplio respaldo teórico (como la teoría de grafos).
Terminología
El término SMILES se refiere a una notación de línea para codificar estructuras moleculares y las instancias específicas deben llamarse estrictamente cadenas SMILES. Sin embargo, el término SONRISAS también se usa comúnmente para referirse tanto a una sola cadena de SONRISAS como a varias cadenas de SONRISAS; el significado exacto suele ser evidente por el contexto. Los términos "canónico" e "isomérico" pueden generar cierta confusión cuando se aplican a SMILES. Los términos describen diferentes atributos de las cadenas SMILES y no se excluyen mutuamente.
Por lo general, se pueden escribir varias cadenas SMILES igualmente válidas para una molécula. Por ejemplo CCO, OCCyC(O)Ctodos especifican la estructura del etanol. Se han desarrollado algoritmos para generar la misma cadena SMILES para una molécula dada; de las muchas cadenas posibles, estos algoritmos eligen solo una de ellas. Este SMILES es único para cada estructura, aunque depende del algoritmo de canonicalización utilizado para generarlo, y se denomina SMILES canónico. Estos algoritmos primero convierten las SONRISAS en una representación interna de la estructura molecular; luego, un algoritmo examina esa estructura y produce una cadena única de SONRISAS. Se han desarrollado varios algoritmos para generar SONRISAS canónicas e incluyen los de Daylight Chemical Information Systems, OpenEye Scientific Software, MEDIT, Chemical Computing Group, MolSoft LLC y el Chemistry Development Kit.
El artículo original que describía el algoritmo CANGEN afirmaba generar cadenas SMILES únicas para gráficos que representan moléculas, pero el algoritmo falla en varios casos simples (p. ej., cuneano, 1,2-diciclopropiletano) y no puede considerarse un método correcto para representar un gráfico. canónicamente Actualmente no existe una comparación sistemática entre el software comercial para probar si tales fallas existen en esos paquetes.
La notación SMILES permite la especificación de la configuración en los centros tetraédricos y la geometría del doble enlace. Estas son características estructurales que no pueden especificarse solo mediante la conectividad y, por lo tanto, las SONRISAS que codifican esta información se denominan SONRISAS isoméricas. Una característica notable de estas reglas es que permiten una especificación parcial rigurosa de la quiralidad. El término SMILES isomérico también se aplica a SMILES en los que se especifican los isómeros.
Definición basada en gráficos
En términos de un procedimiento computacional basado en gráficos, SMILES es una cadena obtenida mediante la impresión de los nodos de símbolos que se encuentran en un recorrido de árbol en profundidad de un gráfico químico. El gráfico químico primero se recorta para eliminar los átomos de hidrógeno y los ciclos se rompen para convertirlo en un árbol de expansión. Cuando se han roto los ciclos, se incluyen etiquetas de sufijos numéricos para indicar los nodos conectados. Los paréntesis se utilizan para indicar puntos de ramificación en el árbol.
La forma SMILES resultante depende de las opciones:
- de los lazos escogidos para romper ciclos,
- del átomo inicial utilizado para el recorrido primero en profundidad, y
- del orden en que se enumeran las ramas cuando se encuentran.
Definición de SMILES como cadenas de un lenguaje libre de contexto
Desde el punto de vista de una teoría formal del lenguaje, SONRISAS es una palabra. A SMILES se puede analizar con un analizador sin contexto. El uso de esta representación ha sido en la predicción de propiedades bioquímicas (incluidas la toxicidad y la biodegradabilidad) basándose en el principio fundamental de la quimioinformática de que moléculas similares tienen propiedades similares. Los modelos predictivos implementaron un enfoque de reconocimiento de patrones sintácticos (que implicó definir una distancia molecular), así como un esquema más sólido basado en el reconocimiento de patrones estadísticos.
Descripción
Átomos
Los átomos están representados por la abreviatura estándar de los elementos químicos, entre corchetes, como el [Au]oro. Los corchetes pueden omitirse en el caso común de átomos que:
- están en el "subconjunto orgánico" de B, C, N, O, P, S, F, Cl, Br o I, y
- no tener cargo formal, y
- tener el número de hidrógenos adjuntos implícito en el modelo de valencia SMILES (típicamente su valencia normal, pero para N y P es 3 o 5, y para S es 2, 4 o 6), y
- son los isótopos normales, y
- no son centros quirales.
Todos los demás elementos deben estar encerrados entre paréntesis y tener cargas e hidrógenos mostrados explícitamente. Por ejemplo, las SONRISAS para el agua pueden escribirse como Oo [OH2]. El hidrógeno también se puede escribir como un átomo separado; El agua también se puede escribir como [H]O[H].
Cuando se utilizan corchetes, se añade el símbolo Hsi el átomo entre paréntesis está unido a uno o más hidrógenos, seguido del número de átomos de hidrógeno si es mayor que 1, luego del signo +de carga positiva o de -carga negativa. Por ejemplo, [NH4+]para amonio (NH4). Si hay más de un cargo, normalmente se escribe como dígito; sin embargo, también es posible repetir el signo tantas veces como el ion tenga cargas: uno puede escribir [Ti+4]o [Ti++++]para titanio(IV) Ti. Así, el anión hidróxido ( OH) está representado por [OH-], el catión hidronio (H 3 O) es [OH3+]y el catión cobalto(III) (Co) es [Co+3]o [Co+++].
Cautiverio
Un bono se representa usando uno de los símbolos . - = # $: / .
Se supone que los enlaces entre átomos alifáticos son simples a menos que se especifique lo contrario y están implícitos por la adyacencia en la cadena SMILES. Aunque los enlaces sencillos se pueden escribir como -, por lo general se omite. Por ejemplo, las SONRISAS para etanol pueden escribirse como C-C-O, CC-Oo C-CO, pero generalmente se escribe CCO.
Los enlaces dobles, triples y cuádruples están representados por los símbolos =, #y $respectivamente, como se ilustra con las SONRISAS O=C=O(dióxido de carbono CO 2), C#N(cianuro de hidrógeno HCN) y [Ga+]$[As-](arseniuro de galio).
Un tipo adicional de enlace es un "no enlace", indicado con ., para indicar que dos partes no están unidas entre sí. Por ejemplo, el cloruro de sodio acuoso se puede escribir [Na+].[Cl-]para mostrar la disociación.
Un enlace aromático "uno y medio" puede indicarse con :; ver § Aromaticidad a continuación.
Los enlaces simples adyacentes a los dobles enlaces pueden representarse usando /o para indicar la configuración estereoquímica; ver § Estereoquímica a continuación.
Anillos
Las estructuras de anillo se escriben rompiendo cada anillo en un punto arbitrario (aunque algunas opciones darán lugar a una SONRISA más legible que otras) para crear una estructura acíclica y agregando etiquetas numéricas de cierre de anillo para mostrar la conectividad entre átomos no adyacentes.
Por ejemplo, ciclohexano y dioxano pueden escribirse como C1CCCCC1y O1CCOCC1respectivamente. Para un segundo anillo, la etiqueta será 2. Por ejemplo, la decalina (decahidronaftaleno) puede escribirse como C1CCCC2C1CCCC2.
SMILES no requiere que los números de timbre se usen en ningún orden en particular y permite el número de timbre cero, aunque rara vez se usa. Además, se permite reutilizar los números de anillo después de que se haya cerrado el primer anillo, aunque esto suele dificultar la lectura de las fórmulas. Por ejemplo, biciclohexilo generalmente se escribe como C1CCCCC1C2CCCCC2, pero también se puede escribir como C0CCCCC0C0CCCCC0.
Múltiples dígitos después de un solo átomo indican múltiples enlaces de cierre de anillo. Por ejemplo, una notación SMILES alternativa para la decalina es C1CCCC2CCCCC12, donde el carbono final participa en los enlaces de cierre de anillo 1 y 2. Si se requieren números de anillo de dos dígitos, la etiqueta está precedida por %, por lo que C%12es un enlace de cierre de anillo simple de anillo 12.
Cualquiera o ambos dígitos pueden estar precedidos por un tipo de enlace para indicar el tipo de enlace de cierre del anillo. Por ejemplo, el ciclopropeno generalmente se escribe C1=CC1, pero si se elige el doble enlace como enlace de cierre del anillo, se puede escribir como C=1CC1, C1CC=1o C=1CC=1. (Se prefiere la primera forma.) C=1CC-1es ilegal, ya que especifica explícitamente tipos en conflicto para el vínculo de cierre del anillo.
Los bonos de cierre de anillo no se pueden usar para denotar bonos múltiples. Por ejemplo, C1C1no es una alternativa válida C=Cpara el etileno. Sin embargo, pueden usarse con no bonos; C1.C2.C12es una forma alternativa peculiar pero legal de escribir propano, más comúnmente escrito CCC.
La elección de un punto de ruptura de anillo adyacente a los grupos adjuntos puede conducir a una forma SMILES más simple al evitar las ramas. Por ejemplo, el ciclohexano-1,2-diol se escribe simplemente como OC1CCCCC1O; elegir una ubicación de ruptura de anillo diferente produce una estructura ramificada que requiere paréntesis para escribir.
Aromaticidad
Los anillos aromáticos como el benceno se pueden escribir en una de tres formas:
- En forma de Kekulé con enlaces simples y dobles alternados, por ejemplo
C1=CC=CC=C1, - Usando el símbolo de enlace aromático
:, por ejemploC1:C:C:C:C:C1, o - Más comúnmente, escribiendo los átomos constituyentes B, C, N, O, P y S en minúsculas
b,c,n, yo, respectivamente.ps
En el último caso, se supone que los enlaces entre dos átomos aromáticos (si no se muestra explícitamente) son enlaces aromáticos. Así, el benceno, la piridina y el furano pueden representarse respectivamente por las SONRISAS c1ccccc1y. n1ccccc1o1cccc1
El nitrógeno aromático unido al hidrógeno, como se encuentra en el pirrol, debe representarse como [nH]; por lo tanto, el imidazol se escribe en notación SMILES como n1c[nH]cc1.
Cuando los átomos aromáticos están unidos entre sí de forma sencilla, como en el bifenilo, un enlace sencillo debe mostrarse explícitamente: c1ccccc1-c2ccccc2. Este es uno de los pocos casos en los que se requiere el símbolo de enlace simple -. (De hecho, la mayoría del software SMILES puede inferir correctamente que el enlace entre los dos anillos no puede ser aromático y aceptará la forma no estándar c1ccccc1c2ccccc2).
Los algoritmos Daylight y OpenEye para generar SONRISAS canónicas difieren en el tratamiento de la aromaticidad.
Derivación
Las ramas se describen entre paréntesis, como en CCC(=O)Oel caso del ácido propiónico y FC(F)Fdel fluoroformo. El primer átomo entre paréntesis y el primer átomo después del grupo entre paréntesis están unidos al mismo átomo de punto de ramificación. El símbolo del bono debe aparecer entre paréntesis; exterior (Ej: CCC=(O)O) no es válido.
Los anillos sustituidos se pueden escribir con el punto de ramificación en el anillo, como se ilustra con las SONRISAS COc(c1)cccc1C#N(ver representación) y COc(cc1)ccc1C#N(ver representación) que codifican los isómeros 3 y 4-cianoanisol. Escribir SONRISAS para los anillos sustituidos de esta manera puede hacerlos más legibles para los humanos.
Las ramas se pueden escribir en cualquier orden. Por ejemplo, bromoclorodifluorometano se puede escribir como FC(Br)(Cl)F, BrC(F)(F)Cl, C(F)(Cl)(F)Bro similar. Generalmente, un formulario SMILES es más fácil de leer si la rama más simple viene primero, siendo la parte final, sin paréntesis, la más compleja. Las únicas advertencias a tales reordenamientos son:
- Si se reutilizan los números de anillo, se emparejan según su orden de aparición en la cadena SMILES. Es posible que se requieran algunos ajustes para preservar el emparejamiento correcto.
- Si se especifica estereoquímica, se deben hacer ajustes; ver Estereoquímica § Notas a continuación.
La única forma de rama que no requiere paréntesis son los enlaces de cierre de anillo. La elección adecuada de enlaces de cierre de anillo puede reducir el número de paréntesis necesarios. Por ejemplo, el tolueno normalmente se escribe como Cc1ccccc1o c1ccccc1C, evitando los paréntesis necesarios si se escribe como c1cc(C)ccc1o c1cc(ccc1)C.
Estereoquímica
SMILES permite, pero no requiere, la especificación de estereoisómeros.
La configuración alrededor de los dobles enlaces se especifica usando los caracteres /y para mostrar enlaces sencillos direccionales adyacentes a un doble enlace. Por ejemplo, F/C=C/F(ver representación) es una representación de trans -1,2-difluoroetileno, en la que los átomos de flúor están en lados opuestos del doble enlace (como se muestra en la figura), mientras que F/C=CF(ver representación) es una posible representación de cis -1,2-difluoroetileno, en el que los flúor están en el mismo lado del doble enlace.
Los símbolos de dirección de enlace siempre vienen en grupos de al menos dos, de los cuales el primero es arbitrario. Es decir, FC=CFes lo mismo que F/C=C/F. Cuando están presentes enlaces simples-dobles alternos, los grupos son más grandes que dos, con los símbolos de dirección central adyacentes a dos enlaces dobles. Por ejemplo, la forma común de (2,4)-hexadieno se escribe C/C=C/C=C/C.
Como ejemplo más complejo, el betacaroteno tiene una columna vertebral muy larga de enlaces simples y dobles alternos, que se puede escribir CC1CCC/C(C)=C1/C=C/C(C)=C/C=C/C(C)=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C2=C(C)/CCCC2(C)C.
La configuración en el carbono tetraédrico se especifica mediante @o @@. Considere los cuatro enlaces en el orden en que aparecen, de izquierda a derecha, en la forma SONRISAS. Mirando hacia el carbono central desde la perspectiva del primer enlace, los otros tres son en sentido horario o antihorario. Estos casos se indican con @@y @, respectivamente (porque el @símbolo en sí es una espiral en sentido contrario a las agujas del reloj).
Por ejemplo, considere el aminoácido alanina. Una de sus formas SONRISAS es NC(C)C(=O)O, más completamente escrita como N[CH](C)C(=O)O. L -alanina, el enantiómero más común, se escribe como N[C@@H](C)C(=O)O(ver representación). Mirando desde el enlace nitrógeno-carbono, los grupos hidrógeno (H), metilo (C) y carboxilato (C(=O)O) aparecen en el sentido de las agujas del reloj. D -Alanina se puede escribir como N[C@H](C)C(=O)O(ver representación).
Si bien el orden en que se especifican las ramas en SMILES normalmente no es importante, en este caso sí lo es; intercambiar dos grupos requiere invertir el indicador de quiralidad. Si las ramas se invierten de modo que alanina se escriba como NC(C(=O)O)C, entonces la configuración también se invierte; L -alanina se escribe como N[C@H](C(=O)O)C(ver representación). Otras formas de escribirlo incluyen C[C@H](N)C(=O)O, OC(=O)[C@@H](N)Cy OC(=O)[C@H](C)N.
Normalmente, el primero de los cuatro enlaces aparece a la izquierda del átomo de carbono, pero si las SONRISAS se escriben comenzando con el carbono quiral, como C(C)(N)C(=O)O, entonces los cuatro están a la derecha, pero el primero en aparecer (el [CH]enlace en este caso) se utiliza como referencia para ordenar los tres siguientes: L -alanina también puede escribirse [C@@H](C)(N)C(=O)O.
La especificación SMILES incluye elaboraciones sobre el @símbolo para indicar estereoquímica alrededor de centros quirales más complejos, como la geometría molecular bipiramidal trigonal.
Isótopos
Los isótopos se especifican con un número igual a la masa isotópica entera que precede al símbolo atómico. El benceno en el que un átomo es carbono-14 se escribe como [14c]1ccccc1y el deuterocloroformo es [2H]C(Cl)(Cl)Cl.
Ejemplos
| Molécula | Estructura | fórmula SONRISAS |
|---|---|---|
| dinitrógeno | N≡N | N#N |
| Isocianato de metilo (MIC) | CH 3 −N=C=O | CN=C=O |
| Sulfato de cobre (II) | CuSO _4 | [Cu+2].[O-]S(=O)(=O)[O-] |
| Vanilina | 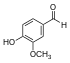 | O=Cc1ccc(O)c(OC)c1COc1cc(C=O)ccc1O |
| Melatonina (C 13 H 16 N 2 O 2) |  | CC(=O)NCCC1=CNc2c1cc(OC)cc2CC(=O)NCCc1c[nH]c2ccc(OC)cc12 |
| Flavopereirina (C 17 H 15 N 2) |  | CCc(c1)ccc2[n+]1ccc3c2[nH]c4c3cccc4CCc1c[n+]2ccc3c4ccccc4[nH]c3c2cc1 |
| Nicotina (C 10 H 14 N 2) |  | CN1CCC[C@H]1c2cccnc2 |
| Enantotoxina (C 17 H 22 O 2) |  | CCC[C@@H](O)CCC=CC=CC#CC#CC=CCOCCC[C@@H](O)CC/C=C/C=C/C#CC#C/C=C/CO |
| Piretrina II (C 22 H 28 O 5) |  | CC1=C(C(=O)C[C@@H]1OC(=O)[C@@H]2[C@H](C2(C)C)/C=C(C)/C(=O)OC)C/C=CC=C |
| Aflatoxina B 1 (C 17 H 12 O 6) | 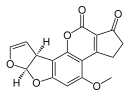 | O1C=C[C@H]([C@H]1O2)c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5 |
| Glucosa (β- D - glucopiranosa) (C 6 H 12 O 6) | 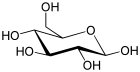 | OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H](O)[C@H](O)1 |
| Bergenina (cuscutina, una resina) (C 14 H 16 O 9) |  | OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H]2[C@@H]1c3c(O)c(OC)c(O)cc3C(=O)O2 |
| Una feromona del insecto escama de California | 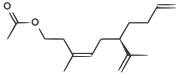 | CC(=O)OCCC(/C)=CC[C@H](C(C)=C)CCC=C |
| (2 S,5 R)-Chalcogran: una feromona del escarabajo descortezador Pityogenes chalcographus | ![(2S,5R)-2-etil-1,6-dioxaespiro[4.4]nonano](https://upload.wikimedia.org/wikipedia/commons/thumb/8/8e/2S%2C5R-chalcogran-skeletal.svg/130px-2S%2C5R-chalcogran-skeletal.svg.png) | CC[C@H](O1)CC[C@@]12CCCO2 |
| α-tujona (C 10 H 16 O) | 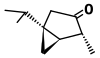 | CC(C)[C@@]12C[C@@H]1[C@@H](C)C(=O)C2 |
| Tiamina (vitamina B 1, C 12 H 17 N 4 OS) | 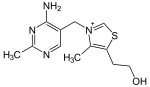 | OCCc1c(C)[n+](cs1)Cc2cnc(C)nc2N |
Para ilustrar una molécula con más de 9 anillos, considere la cefalostatina-1, una pirazina esteroidea de 13 anillos con la fórmula empírica C 54 H 74 N 2 O 10 aislada del hemicordato del Océano Índico Cephalodiscus gilchristi: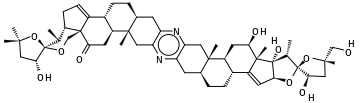
Comenzando con el grupo metilo más a la izquierda en la figura:CC(C)(O1)C[C@@H](O)[C@@]1(O2)[C@@H](C)[C@@H]3CC=C4[C@]3(C2)C(=O)C[C@H]5[C@H]4CC[C@@H](C6)[C@]5(C)Cc(n7)c6nc(C[C@@]89(C))c7C[C@@H]8CC[C@@H]%10[C@@H]9C[C@@H](O)[C@@]%11(C)C%10=C[C@H](O%12)[C@]%11(O)[C@H](C)[C@]%12(O%13)[C@H](O)C[C@@]%13(C)CO
Tenga en cuenta que %aparece delante del índice de las etiquetas de cierre de anillo por encima de 9; ver § Anillos arriba.
Otros ejemplos de SONRISAS
La notación SMILES se describe ampliamente en el manual de teoría SMILES proporcionado por Daylight Chemical Information Systems y se presentan varios ejemplos ilustrativos. La utilidad de representación de Daylight brinda a los usuarios los medios para verificar sus propios ejemplos de SMILES y es una valiosa herramienta educativa.
Extensiones
SMARTS es una notación de línea para la especificación de patrones subestructurales en moléculas. Si bien utiliza muchos de los mismos símbolos que SMILES, también permite la especificación de átomos y enlaces comodín, que se pueden utilizar para definir consultas subestructurales para la búsqueda de bases de datos químicas. Un concepto erróneo común es que la búsqueda subestructural basada en SMARTS implica la coincidencia de cadenas SMILES y SMARTS. De hecho, tanto las cadenas SMILES como SMARTS se convierten primero en representaciones gráficas internas en las que se busca isomorfismo de subgráficos.
SMIRKS, un superconjunto de "SMILES de reacción" y un subconjunto de "SMARTS de reacción", es una notación de línea para especificar transformaciones de reacción. La sintaxis general para las extensiones de reacción es REACTANT>AGENT>PRODUCT(sin espacios), donde cualquiera de los campos puede dejarse en blanco o llenarse con múltiples moléculas delimitadas con un punto (.), y otras descripciones dependen del idioma base. Los átomos también se pueden identificar con un número (por ejemplo [C:1],) para el mapeo, por ejemplo en.
SMILES corresponde a estructuras moleculares discretas. Sin embargo, muchos materiales son macromoléculas, que son demasiado grandes (y a menudo estocásticas) para generar SONRISAS convenientemente. BigSMILES es una extensión de SMILES que tiene como objetivo proporcionar un sistema de representación eficiente para macromoléculas.
Conversión
SMILES se puede volver a convertir en representaciones bidimensionales utilizando algoritmos de generación de diagramas de estructura (SDG). Esta conversión no siempre es inequívoca. La conversión a representación tridimensional se logra mediante enfoques de minimización de energía. Hay muchas utilidades de conversión descargables y basadas en la web.
Contenido relacionado
Semisíntesis
Tabla periódica (estructura cristalina)
Masa molecular