
Síndrome de Stevens-Johnson
La causa más común son ciertos medicamentos como lamotrigina, carbamazepina, alopurinol, antibióticos de sulfonamida y nevirapina. Otras causas pueden incluir infecciones como Mycoplasma pneumoniae y citomegalovirus, o la causa puede permanecer desconocida. Los factores de riesgo incluyen VIH/SIDA y lupus eritematoso sistémico.
El diagnóstico del síndrome de Stevens-Johnson se basa en la afectación de menos del 10 % de la piel. Se conoce como NET cuando afecta a más del 30% de la piel y se considera una forma intermedia cuando afecta al 10-30%. Se cree que las reacciones SJS/TEN siguen un mecanismo de hipersensibilidad de tipo IV. También se incluye con reacción a medicamentos con eosinofilia y síntomas sistémicos (síndrome DRESS), pustulosis exantemática generalizada aguda (PEAG) y necrólisis epidérmica tóxica en un grupo de condiciones conocidas como reacciones adversas cutáneas graves (SCAR).
El tratamiento generalmente se lleva a cabo en un hospital, como una unidad de quemados o una unidad de cuidados intensivos. Los esfuerzos pueden incluir detener la causa, analgésicos, antihistamínicos, antibióticos, inmunoglobulinas intravenosas o corticosteroides. Junto con TEN, SJS afecta a 1 a 2 personas por millón por año. El inicio típico es antes de los 30 años. La piel suele volver a crecer en dos o tres semanas; sin embargo, la recuperación completa puede llevar meses. En general, el riesgo de muerte con SJS es del 5 al 10%.
Signos y síntomas
El SJS generalmente comienza con fiebre, dolor de garganta y fatiga, que comúnmente se diagnostica erróneamente y, por lo tanto, se trata con antibióticos. SJS, SJS/TEN y TEN a menudo son precedidos por fiebre, dolor de garganta, tos y ardor en los ojos durante 1 a 3 días. Los pacientes con estos trastornos experimentan con frecuencia dolor ardiente en la piel al comienzo de la enfermedad. Empiezan a aparecer úlceras y otras lesiones en las mucosas, casi siempre en la boca y los labios, pero también en la región genital y anal. Los que están en la boca suelen ser extremadamente dolorosos y reducen la capacidad del paciente para comer o beber. La conjuntivitis ocurre en alrededor del 30% de los niños que desarrollan SJS. Aparece una erupción de lesiones redondas de aproximadamente una pulgada de ancho en la cara, el tronco, los brazos y las piernas, y las plantas de los pies, pero generalmente no en el cuero cabelludo.
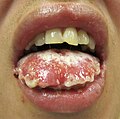
Desquamación de la mucosa en una persona con el síndrome de Stevens-Johnson

Inflamación y peeling de los labios -con llagas que presentan en la lengua y las mucosas en SJS
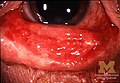
Conjuntivitis en SJS



Causas
Se cree que el SJS surge de un trastorno del sistema inmunitario. La reacción inmunitaria puede desencadenarse por fármacos o infecciones. Los factores genéticos están asociados con una predisposición al SJS. La causa del SJS se desconoce en una cuarta parte a la mitad de los casos. SJS, SJS/TEN y TEN se consideran una sola enfermedad con causas y mecanismos comunes.
Individuos que expresan ciertos serotipos (es decir, alelos genéticos) del antígeno leucocitario humano (es decir, HLA), receptores de células T de base genética o variaciones en su eficiencia para absorber, distribuir a los tejidos, metabolizar o excretar (esta combinación se denomina ADME) un fármaco están predispuestos a desarrollar SJS.
Medicamentos
Aunque el SJS puede ser causado por infecciones virales y tumores malignos, la causa principal son los medicamentos. Una de las principales causas parece ser el uso de antibióticos, en particular las sulfonamidas. Entre 100 y 200 fármacos diferentes pueden estar asociados con SJS. No existe una prueba confiable para establecer un vínculo entre un fármaco en particular y SJS para un caso individual. La determinación de qué fármaco es la causa se basa en el intervalo de tiempo entre el primer uso del fármaco y el comienzo de la reacción cutánea. Es muy poco probable que los medicamentos que se suspenden más de 1 mes antes del inicio de los hallazgos físicos mucocutáneos causen SJS y NET. SJS y TEN comienzan con mayor frecuencia entre 4 y 28 días después de la administración del fármaco culpable. Un algoritmo publicado (ALDEN) para evaluar la causalidad de las drogas brinda asistencia estructurada para identificar la medicación responsable.
SJS puede ser causado por los medicamentos rivaroxabán, vancomicina, alopurinol, valproato, levofloxacina, diclofenaco, etravirina, isotretinoína, fluconazol, valdecoxib, sitagliptina, oseltamivir, penicilinas, barbitúricos, sulfonamidas, fenitoína, azitromicina, oxcarbazepina, zonisamida, modafinilo, lamotrigina, nevirapina, pirimetamina, ibuprofeno, etosuximida, carbamazepina, bupropión, telaprevir y nistatina.
Los medicamentos que tradicionalmente se ha sabido que provocan SJS, eritema multiforme y necrólisis epidérmica tóxica incluyen antibióticos de sulfonamida, antibióticos de penicilina, cefixima (antibiótico), barbitúricos (sedantes), lamotrigina, fenitoína (p. ej., Dilantin) (anticonvulsivos) y trimetoprima La combinación de lamotrigina con valproato de sodio aumenta el riesgo de SJS.
Los medicamentos antiinflamatorios no esteroideos (NSAID, por sus siglas en inglés) son una causa rara de SJS en adultos; el riesgo es mayor para los pacientes mayores, las mujeres y los que inician el tratamiento. Por lo general, los síntomas del SJS inducido por medicamentos surgen dentro de la semana posterior al inicio del medicamento. Al igual que los AINE, el paracetamol (acetaminofén) también ha causado casos raros de SJS. Las personas con lupus eritematoso sistémico o infecciones por VIH son más susceptibles al SJS inducido por fármacos.
Infecciones
La segunda causa más común de SJS y TEN es la infección, particularmente en los niños. Esto incluye infecciones de las vías respiratorias superiores, otitis media, faringitis e infecciones por el virus de Epstein-Barr, Mycoplasma pneumoniae y citomegalovirus. El uso rutinario de medicamentos como antibióticos, antipiréticos y analgésicos para controlar las infecciones puede dificultar la identificación de si los casos fueron causados por la infección o por los medicamentos que se tomaron.
Las enfermedades virales que causan SJS incluyen: el virus del herpes simple (posiblemente; se debate), el SIDA, el virus coxsackie, la influenza, la hepatitis y las paperas.
En casos pediátricos, el virus de Epstein-Barr y los enterovirus se han asociado con el SJS.
Más de la mitad de los pacientes con SSJ han notificado infecciones recientes de las vías respiratorias superiores.
Las infecciones bacterianas relacionadas con el SSJ incluyen estreptococos beta-hemolíticos del grupo A, difteria, brucelosis, linfogranuloma venéreo, micobacterias, Mycoplasma pneumoniae, infecciones por rickettsiosis, tularemia y fiebre tifoidea.
Las infecciones fúngicas con coccidioidomicosis, dermatofitosis e histoplasmosis también se consideran posibles causas. La malaria y la tricomoniasis, infecciones por protozoos, también se han informado como causas.
Fisiopatología
SJS es una reacción de hipersensibilidad de tipo IV en la que un fármaco o su metabolito estimula las células T citotóxicas (es decir, células T CD8+) y células T auxiliares (es decir, CD4+ células T) para iniciar reacciones autoinmunes que atacan los tejidos propios. En particular, es una reacción de hipersensibilidad retardada de tipo IV, subtipo IVc, que depende en parte de las acciones de daño tisular de las células asesinas naturales. Esto contrasta con los otros tipos de trastornos SCAR, es decir, el síndrome DRESS, que es una reacción de hipersensibilidad al fármaco de tipo IV, subtipo IVb, dependiente en parte de las acciones lesionadoras de los tejidos de los eosinófilos, y la pustulosis exantemática generalizada aguda, que es un subtipo de tipo IV. IVd, reacción de hipersensibilidad dependiente en parte de las acciones de daño tisular de los neutrófilos.
Al igual que otros fármacos inductores de SCAR, los fármacos inductores de SJS o sus metabolitos estimulan las células T CD8+ o las células T CD4+ para iniciar respuestas autoinmunes. Los estudios indican que el mecanismo por el cual un fármaco o sus metabolitos logran esto implica subvertir las vías de presentación de antígenos del sistema inmunitario innato. El fármaco o metabolito se une covalentemente con una proteína huésped para formar un epítopo no propio relacionado con el fármaco. Una célula presentadora de antígenos (APC) toma estas proteínas alteradas; los digiere en pequeños péptidos; coloca los péptidos en un surco en el componente del antígeno leucocitario humano (es decir, HLA) de su complejo principal de histocompatibilidad (es decir, MHC); y presenta los péptidos asociados al MHC a los receptores de células T en las células T CD8+ o en las células T CD4+. Los péptidos que expresan un epítopo no propio relacionado con el fármaco en una de sus diversas formas de proteína HLA (HLA-A, HLA-B, HLA-C, HLA-DM, HLA-DO, HLA-DP, HLA-DQ o HLA-DR) puede unirse a un receptor de células T y, por lo tanto, estimular a la célula T progenitora portadora del receptor para que inicie ataques en los propios tejidos. Alternativamente, un fármaco o su metabolito pueden estimular estas células T al insertarse en el surco de una proteína HLA para servir como un epítopo no propio o unirse fuera de este surco para alterar una proteína HLA para que forme un epítopo no propio. Sin embargo, en todos estos casos, un epítopo no propio debe unirse a un serotipo HLA específico (es decir, una variación) para estimular las células T. Dado que la población humana expresa unos 13 000 serotipos HLA diferentes, mientras que un individuo expresa solo una fracción de ellos y dado que un fármaco o metabolito inductor del SJS interactúa con solo uno o unos pocos serotipos HLA, la capacidad de un fármaco para inducir SCAR es limitada. a aquellas personas que expresan serotipos HLA a los que se dirige el fármaco o su metabolito. En consecuencia, solo unas pocas personas están predispuestas a desarrollar SCAR en respuesta a un medicamento en particular sobre la base de su expresión de serotipos HLA: Los estudios han identificado varios serotipos HLA asociados con el desarrollo de SJS, SJS/TEN o TEN en respuesta a ciertos medicamentos.. En general, estas asociaciones se restringen a las poblaciones citadas.
En algunas poblaciones de Asia oriental estudiadas (chinos Han y tailandeses), el SJS inducido por carbamazepina y fenitoína está fuertemente asociado con HLA-B*1502 (HLA-B75), un serotipo HLA-B del serotipo más amplio HLA-B15. Un estudio en Europa sugirió que el marcador genético solo es relevante para los asiáticos orientales. Esto tiene relevancia clínica ya que se acordó que antes de iniciar un medicamento como el alopurinol en un paciente de ascendencia china, se debe considerar la prueba HLA-B*58:01.
Según los hallazgos asiáticos, estudios similares en Europa mostraron que el 61 % de los pacientes con SJS/NET inducidos por alopurinol portaban el HLA-B58 (la frecuencia del fenotipo del alelo B*5801 en los europeos suele ser del 3 %). Un estudio concluyó: "Incluso cuando los alelos HLA-B se comportan como fuertes factores de riesgo, como ocurre con el alopurinol, no son ni suficientes ni necesarios para explicar la enfermedad".
Otras asociaciones de HLA con el desarrollo de SJS, SJS/TEN o TEN y la ingesta de medicamentos específicos determinados en ciertas poblaciones se dan en las asociaciones de HLA con SCAR.
Receptores de células T
Además de actuar a través de las proteínas HLA para unirse con un receptor de células T, un fármaco o su metabolito puede pasar por alto las proteínas HLA para unirse directamente a un receptor de células T y, por lo tanto, estimular la T CD8+. o células T CD4+ para iniciar respuestas autoinmunes. En cualquier caso, esta unión parece desarrollarse solo en ciertos receptores de células T. Dado que los genes para estos receptores están muy modificados, es decir, alterados para codificar proteínas con diferentes secuencias de aminoácidos, y dado que la población humana puede expresar más de 100 billones de receptores de células T diferentes (es decir, secuencias de aminoácidos diferentes), mientras que un individuo expresa solo un fracción de estos, la capacidad de un fármaco o de su metabolito para inducir el síndrome de DRESS al interactuar con un receptor de células T se limita a aquellos individuos cuyas células T expresan uno o más receptores de células T que pueden interactuar con la droga o su metabolito. Por lo tanto, solo individuos raros están predispuestos a desarrollar SJS en respuesta a un fármaco particular sobre la base de su expresión de tipos de receptores de células T específicos. Si bien la evidencia que respalda esta selectividad del receptor de células T es limitada, un estudio identificó la presencia preferencial de TCR-V-b y la región 3 determinante de la complementariedad en los receptores de células T que se encuentran en las células T en las ampollas de pacientes con DRESS inducido por alopurinol. síndrome. Este hallazgo es compatible con la noción de que tipos específicos de receptores de células T están involucrados en el desarrollo de SCAR específicas inducidas por fármacos.
ADME
Se han encontrado variaciones en ADME, es decir, la eficiencia de un individuo para absorber, distribuir tejidos, metabolizar o excretar un fármaco, en diversas reacciones adversas cutáneas graves (SCARS), así como en otros tipos de reacciones adversas. reacciones a medicamentos Estas variaciones influyen en los niveles y la duración de un fármaco o su metabolito en los tejidos y, por lo tanto, afectan la capacidad del fármaco o del metabolito para provocar estas reacciones. Por ejemplo, CYP2C9 es un importante citocromo P450 que metaboliza fármacos; metaboliza y por lo tanto inactiva la fenitoína. Las personas de Taiwán, Japón y Malasia que expresan la variante CYP2C9*3 de CYP2C9, que tiene una actividad metabólica reducida en comparación con el citocromo de tipo salvaje (es decir, CYP2c9*1), tienen niveles sanguíneos elevados de fenitoína y una alta incidencia de SJS (así como SJS/TEN y TEN) al tomar el medicamento. Además de las anomalías en las enzimas metabolizadoras de fármacos, se sugiere que las disfunciones del riñón, el hígado o el tracto GI que aumentan los niveles de un fármaco o metabolito inductor de SCAR promueven las respuestas de SCAR. Estas anormalidades de ADME, también se sugiere, pueden interactuar con proteínas HLA particulares y receptores de células T para promover un trastorno SCAR.
Diagnóstico
El diagnóstico se basa en la afectación de menos del 10 % de la piel. Se conoce como TEN cuando se afecta más del 30% de la piel y una forma intermedia con una afectación del 10 al 30%. Un signo de Nikolsky positivo es útil en el diagnóstico de SJS y NET. Una biopsia de piel es útil, pero no obligatoria, para establecer un diagnóstico de SJS y NET.
Patología

SJS, como NET y eritema multiforme, se caracteriza por necrosis epidérmica confluente con mínima inflamación asociada. La agudeza es evidente por el patrón (normal) en forma de tejido de canasta del estrato córneo.
Clasificación
El síndrome de Stevens-Johnson (SSJ) es una forma más leve de necrólisis epidérmica tóxica (TEN). Estas condiciones se reconocieron por primera vez en 1922. Una clasificación publicada por primera vez en 1993, que se adoptó como una definición de consenso, identifica el síndrome de Stevens-Johnson, la necrólisis epidérmica tóxica y la superposición SJS/NET. Los tres forman parte de un espectro de reacciones cutáneas graves (SCAR) que afectan a la piel y las mucosas. La distinción entre SJS, superposición SJS/TEN y TEN se basa en el tipo de lesiones y la cantidad de área de superficie corporal con ampollas y erosiones. Se acuerda que el método más confiable para clasificar EM, SJS y NET se basa en la morfología de la lesión y la extensión del desprendimiento epidérmico. Las ampollas y las erosiones cubren entre el 3 % y el 10 % del cuerpo en SJS, entre el 11 y el 30 % en la superposición SJS/NET y más del 30 % en TEN. El patrón de piel más comúnmente asociado con SJS es generalizado, a menudo unido o en contacto (confluente), manchas papúricas (máculas) o pequeñas ampollas planas o ampollas grandes que también pueden unirse. Estos ocurren principalmente en el torso.
La superposición de SJS, TEN y SJS/TEN puede confundirse con eritema multiforme. El eritema multiforme, que también está dentro del espectro SCAR, difiere en el patrón clínico y la etiología.
Prevención
Se recomienda o está en estudio evaluar a las personas para detectar ciertas variantes genéticas predisponentes antes de iniciar el tratamiento con determinados fármacos inductores de SJS, TEN/SJS o TEN. Estas recomendaciones generalmente se limitan a poblaciones específicas que muestran una posibilidad significativa de tener la variante genética indicada, ya que la detección de poblaciones con incidencias extremadamente bajas de expresión de la variante se considera rentable. Las personas que expresan el alelo HLA asociado con la sensibilidad a un fármaco indicado no deben recibir tratamiento con el fármaco. Estas recomendaciones incluyen lo siguiente. Antes del tratamiento con carbamazepina, las Administraciones de Alimentos y Medicamentos de Taiwán y EE. UU. recomiendan la detección de HLA-B*15:02 en ciertos grupos asiáticos. Esto se ha implementado en Taiwán, Hong Kong, Singapur y muchos centros médicos en Tailandia y China continental. Antes del tratamiento con alopurinol, las pautas del American College of Rheumatology para el manejo de la gota recomiendan la detección de HLA-B*58:01. Esto se proporciona en muchos centros médicos en Taiwán, Hong Kong, Tailandia y China continental. Antes del tratamiento con abacavir, la Administración de Drogas y Alimentos de EE. UU. recomienda la detección de HLA-B*57:01 en poblaciones caucásicas. Esta evaluación está ampliamente implementada. También se ha sugerido que todas las personas que expresen este serotipo HLA eviten el tratamiento con abacovir. Se están realizando ensayos actuales en Taiwán para definir la rentabilidad de evitar la fenitoína en SJS, SJS/TEN y TEN para personas que expresan el alelo CYP2C9*3 de CYP2C9.
Tratamiento
El SSJ constituye una urgencia dermatológica. Los pacientes con infecciones documentadas por Mycoplasma pueden tratarse con macrólidos orales o doxiciclina oral.
Al principio, el tratamiento es similar al de los pacientes con quemaduras térmicas, y la atención continua solo puede ser de apoyo (p. ej., líquidos intravenosos y alimentación nasogástrica o parenteral) y sintomática (p. ej., enjuague bucal analgésico para las úlceras bucales). Los dermatólogos y los cirujanos tienden a estar en desacuerdo acerca de si la piel debe desbridarse.
Más allá de este tipo de atención de apoyo, no se acepta ningún tratamiento para el SJS. El tratamiento con corticoides es controvertido. Los primeros estudios retrospectivos sugirieron que los corticosteroides aumentaron las estancias hospitalarias y las tasas de complicaciones. No se han realizado ensayos aleatorios de corticosteroides para SJS, y se puede manejar con éxito sin ellos.
Se han utilizado otros agentes, como la ciclofosfamida y la ciclosporina, pero ninguno ha tenido mucho éxito terapéutico. El tratamiento con inmunoglobulina intravenosa se ha mostrado prometedor para reducir la duración de la reacción y mejorar los síntomas. Otras medidas de apoyo comunes incluyen el uso de anestésicos y antisépticos tópicos para el dolor, mantener un ambiente cálido y analgésicos intravenosos.
Se debe consultar a un oftalmólogo de inmediato, ya que el SJS con frecuencia causa la formación de tejido cicatricial dentro de los párpados, lo que lleva a la vascularización de la córnea, problemas de visión y muchos otros problemas oculares. Aquellos con enfermedad crónica de la superficie ocular causada por SJS pueden encontrar alguna mejoría con el tratamiento PROSE (tratamiento de reemplazo protésico del ecosistema de la superficie ocular).
Pronóstico
SJS (con menos del 10 % de la superficie corporal afectada) tiene una tasa de mortalidad de alrededor del 5 %. La mortalidad por necrólisis epidérmica tóxica (TEN) es del 30 al 40%. El riesgo de muerte se puede estimar mediante la escala SCORTEN, que tiene en cuenta una serie de indicadores pronósticos. Es útil calcular un SCORTEN dentro de los primeros 3 días de hospitalización. Otros resultados incluyen daño/fallo de órganos, rascado de la córnea y ceguera. Se puede desarrollar enfermedad pulmonar restrictiva en pacientes con SJS y NET después de la afectación pulmonar aguda inicial. Los pacientes con SJS o NET causados por un fármaco tienen mejor pronóstico cuanto antes se retire el fármaco causante.
Epidemiología
SJS es una condición rara, con una incidencia reportada de alrededor de 2,6 a 6,1 casos por millón de personas por año. En los Estados Unidos, cada año se realizan alrededor de 300 nuevos diagnósticos. La condición es más común en adultos que en niños.
Historia
SJS lleva el nombre de Albert Mason Stevens y Frank Chambliss Johnson, pediatras estadounidenses que publicaron conjuntamente una descripción del trastorno en el American Journal of Diseases of Children en 1922.
Casos destacados
- Ab-Soul, American hip hop recording artist and member of Black Hippy
- Padma Lakshmi, actriz, modelo, personalidad de la televisión y escritor de libros de cocina
- Manute Bol, ex jugador de NBA. Bol murió de complicaciones del síndrome de Stevens-Johnson, así como de insuficiencia renal.
- Gene Sauers, tres veces PGA Ganador del Tour
- Samantha Reckis, una niña de siete años Plymouth, Massachusetts que perdió la piel cubriendo el 95% de su cuerpo después de tomar Motrin infantil en 2003. En 2013, un jurado le otorgó $63M en una demanda contra Johnson & Johnson, una de las mayores demandas de su tipo. La decisión fue confirmada en 2015.
- Karen Elaine Morton, modelo y actriz que apareció en el video "867-5309/Jenny" de Tommy Tutone y fue Playmate del Mes en el número de julio de 1978 Playboy Revista.
Investigación
En 2015, los NIH y la Administración de Alimentos y Medicamentos (FDA) organizaron un taller titulado "Direcciones de investigación en el síndrome de Stevens-Johnson mediado genéticamente/necrólisis epidérmica tóxica".
Contenido relacionado
Malaria
Manía
Administración de Alimentos y Medicamentos