
Principio de Curtin-Hammett
El principio de Curtin-Hammett es un principio de cinética química propuesto por David Yarrow Curtin y Louis Plack Hammett. Establece que, para una reacción que tiene un par de intermedios reactivos o reactivos que se interconvierten rápidamente (como suele ser el caso de los isómeros conformacionales), cada uno de los cuales se dirige irreversiblemente a un producto diferente, la relación de productos dependerá tanto de la diferencia de energía entre los dos confórmeros y las barreras de energía de cada uno de los isómeros que se equilibran rápidamente a sus respectivos productos. Dicho de otra manera, la distribución del producto refleja la diferencia de energía entre los dos estados de transición que limitan la velocidad. Como resultado, la distribución del producto no necesariamente reflejará la distribución de equilibrio de los dos intermedios.Se ha invocado el principio de Curtin-Hammett para explicar la selectividad en una variedad de reacciones estereo y regioselectivas. La relación entre las constantes de velocidad (aparentes) y la constante de equilibrio se conoce como la ecuación de Winstein-Holness.
Definición
El principio de Curtin-Hammett se aplica a sistemas en los que se forman diferentes productos a partir de dos sustratos en equilibrio entre sí. Los reactivos que se interconvierten rápidamente pueden tener cualquier relación entre ellos (estereoisómeros, isómeros constitucionales, isómeros conformacionales, etc.). La formación del producto debe ser irreversible, y los diferentes productos deben ser incapaces de interconvertirse.
Por ejemplo, dadas las especies A y B que se equilibran rápidamente mientras que A se convierte irreversiblemente en C y B se convierte irreversiblemente en D:
![{displaystyle {ce {bf {{C} {it {<-[k_{rm {1}}]{bf {{A}{it { <=>[{K}] {bf {{B} {it {->[k_{rm {2}}] {bf {D}}}}}}}}}}}}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8bf279b211853c9be67aece08ca6504276d4b50d)
K es la constante de equilibrio entre A y B, y k 1 y k 2 son las constantes de velocidad para la formación de C y D, respectivamente. Cuando la tasa de interconversión entre A y B es mucho más rápida que k 1 o k 2, entonces el principio de Curtin-Hammett nos dice que la relación del producto C: D no es igual al equilibrio A: Brelación de reactivos, sino que está determinada por las energías relativas de los estados de transición (es decir, la diferencia en las energías absolutas de los estados de transición). Si los reactivos A y B tuvieran energías idénticas, la relación de productos dependería solo de las barreras de activación de las reacciones que conducen a cada producto respectivo. Sin embargo, en un escenario del mundo real, es probable que los dos reactivos tengan niveles de energía algo diferentes, aunque la barrera para su interconversión debe ser baja para que se aplique el escenario de Curtin-Hammett. En este caso, la distribución del producto depende tanto de la relación de equilibrio de A a B como de las barreras de activación relativas que van a los productos correspondientes C yD. _ Ambos factores se tienen en cuenta por la diferencia de energías de los estados de transición (ΔΔ G en la figura siguiente).
El perfil de energía libre de coordenadas de reacción de una reacción típica bajo el control de Curtin-Hammett se representa mediante la siguiente figura:
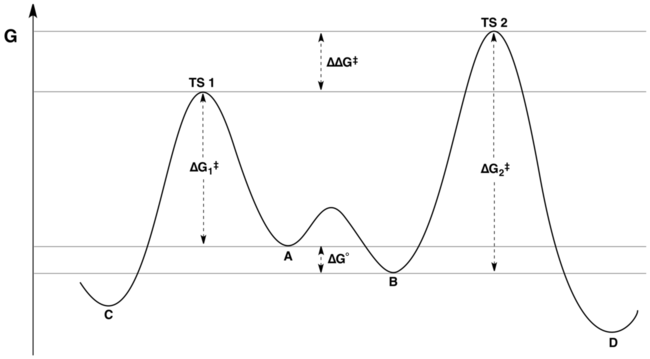
La proporción de productos solo depende del valor etiquetado como ΔΔ G en la figura: C será el producto principal, porque la energía de TS1 es menor que la energía de TS2. Una afirmación común pero falsa es que la distribución del producto no refleja de ninguna manera las energías libres relativas de los sustratos A y B; de hecho, refleja las energías libres relativas de los sustratos y las energías de activación relativas.Este malentendido puede provenir de no apreciar la distinción entre "la diferencia de energías de activación" y "la diferencia en las energías del estado de transición". Aunque estas cantidades pueden parecer sinónimas al principio, la segunda tiene en cuenta la constante de equilibrio para la interconversión de A y B, mientras que la primera no.
Matemáticamente, la relación del producto se puede expresar como una función de K, k 1 y k 2 o en términos de las energías correspondientes Δ G °, Δ G 1 y Δ G 2. Al combinar términos, la relación del producto se puede reescribir en términos de la cantidad ΔΔ G solo, donde ΔΔ G = (Δ G 2 – Δ G 1) + Δ G °. La inspección del diagrama de energía (que se muestra arriba) hace evidente que ΔΔ G es precisamente la diferencia en las energías del estado de transición.
Derivación
Una reacción genérica bajo Curtin-Hammett se puede describir mediante los siguientes parámetros:
Para que el equilibrio rápido sea una buena suposición, la tasa de conversión de A o B menos estable al producto C o D debe ser al menos 10 veces más lenta que la tasa de equilibrio entre A y B.
La velocidad de formación del compuesto C a partir de A se da como![{displaystyle {frac {d[mathbf {C} ]}{dt}}=k_{1}[mathbf {A} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7a0ba64b7c5b396fc651fb3aad19d80574dcdcdc)
y el de D de B como![{displaystyle {frac {d[mathbf {D} ]}{dt}}=k_{2}[mathbf {B} ]approx k_{2}K[mathbf {A} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e28151c0c25112e0df3d966f7dd520e4e3071c7f)
con la segunda igualdad aproximada que sigue de la suposición de equilibrio rápido. Bajo esta suposición, la relación de los productos es entonces![{displaystyle {frac {[mathbf {D} ]}{[mathbf {C} ]}}approx {frac {d[mathbf {D} ]}{dt}}{Big /}{ frac {d[mathbf {C} ]}{dt}}={frac {k_{2}[mathbf {B} ]}{k_{1}[mathbf {A} ]}}approx { frac {k_{2}K[mathbf {A} ]}{k_{1}[mathbf {A} ]}}={frac {k_{2}K}{k_{1}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/802efbeb75055ff826c7417118672230e54b99a2)
En otras palabras, porque el equilibrio es rápido en comparación con la formación del producto, ![{displaystyle [mathbf {B} ]/[mathbf {A} ]aprox. K}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e2f3778641d32c658e4c1ab2961f8735a102b329)
![{displaystyle {frac {d[mathbf {D} ]}{dt}}{Big /}{frac {d[mathbf {C} ]}{dt}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4f5d950d8350d10e6de7d872eea6986bbdd08b21)
![{displaystyle [mathbf {D} ]/[mathbf {C} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/89143f8cee66e6e3cc5c89dea98fc4b2d65c8bc0)
En términos de las energías del estado fundamental y del estado de transición, la relación del producto se puede escribir como:![{displaystyle {frac {[mathbf {D} ]}{[mathbf {C} ]}}approx {frac {k_{2}K}{k_{1}}}={frac {e ^{-Delta G_{2}^{ddagger }/RT}e^{-Delta G^{circ }/RT}}{e^{-Delta G_{1}^{ddagger }/ RT}}}=exp {grande (}-(Delta G_{2}^{ddagger }-Delta G_{1}^{ddagger }+Delta G^{circ })/RT{ grande)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b18fc40f30584687fab6672e87c8c07bc0160936)
Es importante destacar que la inspección del diagrama de energía anterior nos permite escribir
dándonos una ecuación simplificada que captura la esencia del principio de Curtin-Hammett:
![{displaystyle {frac {[mathbf {D} ]}{[mathbf {C} ]}}approx e^{-Delta Delta G^{ddagger}/RT}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0d4bbae9e76a67c396d9d0cc15dc87bab82b7c6)
Por tanto, aunque la relación del producto depende de la constante de equilibrio entre A y B y de la diferencia de energía entre las barreras de A a C y de B a D, ambos factores se tienen en cuenta automáticamente por la diferencia de energía de los estados de transición. dando lugar a los productos, ΔΔ G.
Clases de reacciones bajo el control de Curtin-Hammett
El principio de Curtin-Hammett puede explicar tres clases principales de reacciones: el confórmero más o menos estable puede reaccionar más rápidamente, o ambos pueden reaccionar a la misma velocidad.
Caso I: el confórmero más estable reacciona más rápidamente
Una categoría de reacciones bajo el control de Curtin-Hammett incluye transformaciones en las que el confórmero más estable reacciona más rápidamente. Esto ocurre cuando el estado de transición del intermediario mayoritario a su respectivo producto tiene menor energía que el estado de transición del intermediario menor al otro posible producto. Entonces, el producto principal se deriva del confórmero principal, y la distribución del producto no refleja la distribución del confórmero de equilibrio.
Ejemplo: oxidación de piperidina
Un ejemplo de un escenario de Curtin-Hammett en el que el isómero conformacional más estable reacciona más rápidamente se observa durante la oxidación de piperidinas. En el caso de la N-metilpiperidina, la inversión en nitrógeno entre confórmeros diastereoisómeros es mucho más rápida que la velocidad de oxidación de la amina. La conformación que coloca al grupo metilo en la posición ecuatorial es 3,16 kcal/mol más estable que la conformación axial. La relación de producto de 95:5 indica que el confórmero más estable conduce al producto principal.

Caso II: el confórmero menos estable reacciona más rápidamente
Una segunda categoría de reacciones bajo el control de Curtin-Hammett incluye aquellas en las que el confórmero menos estable reacciona más rápidamente. En este caso, a pesar de una preferencia energética por las especies menos reactivas, el producto principal se deriva de las especies de mayor energía. Una implicación importante es que el producto de una reacción puede derivarse de un confórmero que se encuentra en una concentración lo suficientemente baja como para ser inobservable en el estado fundamental.
Ejemplo: alquilación de tropano
La alquilación de tropanos con yoduro de metilo es un ejemplo clásico de un escenario de Curtin-Hammett en el que puede surgir un producto principal de una conformación menos estable. Aquí, el confórmero menos estable reacciona a través de un estado de transición más estable para formar el producto principal. Por lo tanto, la distribución conformacional del estado fundamental no refleja la distribución del producto.

Caso III: ambos confórmeros reaccionan a la misma velocidad
Es hipotéticamente posible que dos confórmeros diferentes en equilibrio puedan reaccionar a través de estados de transición que son iguales en energía. En este caso, la selectividad del producto dependería únicamente de la distribución de los confórmeros del estado fundamental. En este caso, ambos confórmeros reaccionarían a la misma velocidad.
Ejemplo: reacción S N 2 de yoduro de ciclohexilo
Ernest L. Eliel ha propuesto que la reacción hipotética del yoduro de ciclohexilo con el yoduro radiomarcado daría como resultado un estado de transición completamente simétrico. Debido a que los confórmeros ecuatoriales y axialmente sustituidos reaccionarían a través del mismo estado de transición, ΔΔG sería igual a cero. Por el principio de Curtin-Hammett, la distribución de productos debe ser entonces 50% axial sustituida y 50% ecuatorial sustituida. Sin embargo, el equilibrio de los productos impide la observación de este fenómeno.

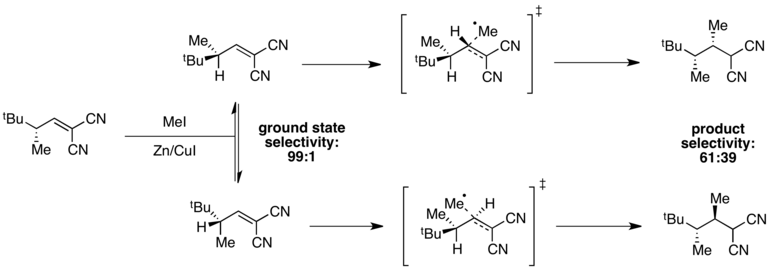
Ejemplo: metilación por radicales
Cuando las energías del estado fundamental son diferentes pero las energías del estado de transición son similares, la selectividad se degradará en el estado de transición y se puede observar una selectividad general deficiente. Por ejemplo, se observa una alta selectividad por un confórmero en estado fundamental en la siguiente reacción de metilación por radicales.

El confórmero en el que se minimiza la deformación A(1,3) tiene un mínimo de energía, lo que proporciona una selectividad de 99:1 en el estado fundamental. Sin embargo, las energías del estado de transición dependen tanto de la presencia de la tensión A(1,3) como del impedimento estérico asociado con el radical metilo entrante. En este caso, estos dos factores están en oposición y la diferencia en las energías del estado de transición es pequeña en comparación con la diferencia en las energías del estado fundamental. Como resultado, se observa una escasa selectividad global en la reacción.
Aplicación a reacciones estereoselectivas y regioselectivas
El principio de Curtin-Hammett se utiliza para explicar las relaciones de selectividad de algunas reacciones estereoselectivas.
Aplicación a la resolución cinética dinámica
El principio de Curtin-Hammett puede explicar la dinámica observada en transformaciones que emplean resolución cinética dinámica, como la hidrogenación asimétrica de Noyori y la litiación enantioselectiva.
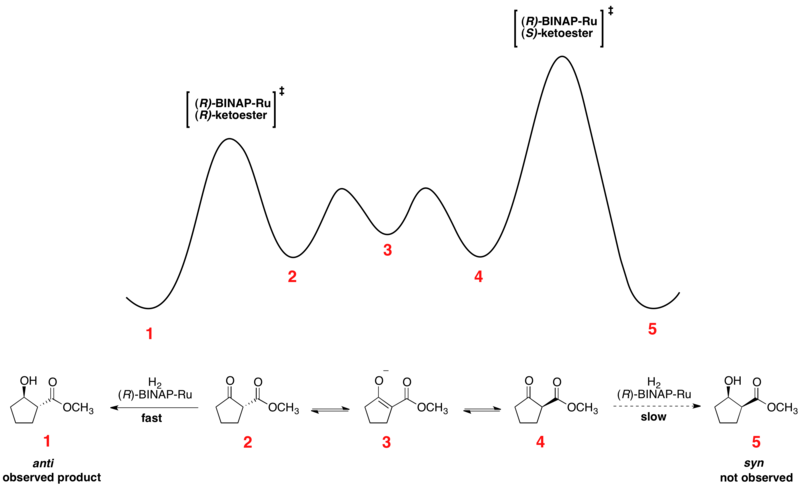
Hidrogenación asimétrica de Noyori
El rápido equilibrio entre los confórmeros enantioméricos y la hidrogenación irreversible colocan la reacción bajo el control de Curtin-Hammett. El uso de un catalizador quiral da como resultado un estado de transición de mayor y menor energía para la hidrogenación de los dos enantiómeros. La transformación ocurre a través del estado de transición de menor energía para formar el producto como un solo enantiómero. De acuerdo con el principio de Curtin-Hammett, la proporción de productos depende de la barrera energética absoluta del paso irreversible de la reacción y no refleja la distribución de equilibrio de los confórmeros del sustrato. El perfil de energía libre relativa de un ejemplo de hidrogenación asimétrica de Noyori se muestra a continuación:
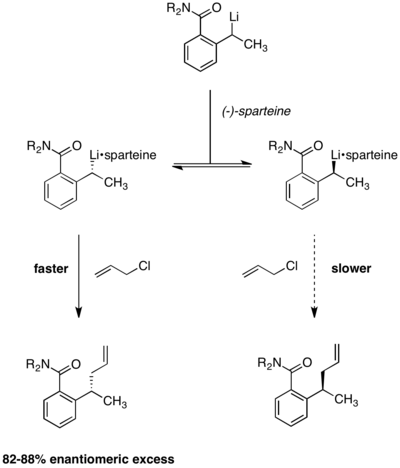
Litiación enantioselectiva
La resolución cinética dinámica en condiciones de Curtin-Hammett también se ha aplicado a las reacciones de litiación enantioselectiva. En la siguiente reacción, se observó que las enantioselectividades de los productos eran independientes de la quiralidad del material de partida. El uso de (-)-esparteína es esencial para la enantioselectividad, formándose un producto racémico en su ausencia. El equilibrio entre los dos complejos de alquillitio se demostró mediante la observación de que la enantioselectividad permaneció constante durante el transcurso de la reacción. Si los dos complejos reactivos no se interconvirtieran rápidamente, la enantioselectividad se erosionaría con el tiempo a medida que se agotara el confórmero de reacción más rápida.
Aplicación a la acilación regioselectiva
Se ha invocado el principio de Curtin-Hammett para explicar la regioselectividad en la acilación de 1,2-dioles. Normalmente, el sitio menos impedido de un 1,2-diol asimétrico experimentaría una esterificación más rápida debido a la reducción del impedimento estérico entre el diol y el reactivo acilante. Desarrollar una esterificación selectiva del grupo hidroxilo más sustituido es una transformación útil en química orgánica sintética, particularmente en la síntesis de carbohidratos y otros compuestos polihidroxilados. Se han utilizado acetales de estannileno para lograr esta transformación de manera eficiente.

El diol asimétrico se trata primero con un reactivo de estaño para producir el acetal de dibutilestannileno. Luego, este compuesto se trata con un equivalente de cloruro de acilo para producir el monoéster de estannilo. Dos isómeros del éster de estannilo son accesibles y pueden sufrir una rápida interconversión a través de un intermedio tetraédrico. Inicialmente, predomina el isómero menos estable, ya que se forma más rápidamente a partir del acetal de estannilo. Sin embargo, permitir que los dos isómeros se equilibren da como resultado un exceso del alcoxistanano primario más estable en solución. A continuación, la reacción se detiene irreversiblemente, reaccionando más rápidamente el alcoxistanano primario menos impedido. Esto da como resultado la producción selectiva del monoéster más sustituido. Este es un escenario de Curtin-Hammett en el que el isómero más estable también reacciona más rápidamente.

Aplicación a la epoxidación asimétrica
La epoxidación de alquenos asimétricos también se ha estudiado como un ejemplo de la cinética de Curtin-Hammett. En un estudio computacional de la epoxidación diastereoselectiva de alcoholes alílicos quirales por complejos de peroxi de titanio, la diferencia calculada en las energías del estado de transición entre los dos confórmeros fue de 1,43 kcal/mol. Experimentalmente, la relación de producto observada fue de 91:9 a favor del producto derivado del estado de transición de menor energía. Esta relación de productos es consistente con la diferencia calculada en las energías del estado de transición. Este es un ejemplo en el que el confórmero favorecido en el estado fundamental, que experimenta una deformación A(1,3) reducida, reacciona a través de un estado de transición de menor energía para formar el producto principal.

Aplicaciones sintéticas
Síntesis de AT2433-A1
Se ha invocado el principio de Curtin-Hammett para explicar la selectividad en una variedad de vías sintéticas. Se observa un ejemplo en el camino hacia el antibiótico antitumoral AT2433-A1, en el que se produce una ciclación de tipo Mannich con excelente regioselectividad. Los estudios demuestran que el paso de ciclación es irreversible en el solvente utilizado para ejecutar la reacción, lo que sugiere que la cinética de Curtin-Hammett puede explicar la selectividad del producto.

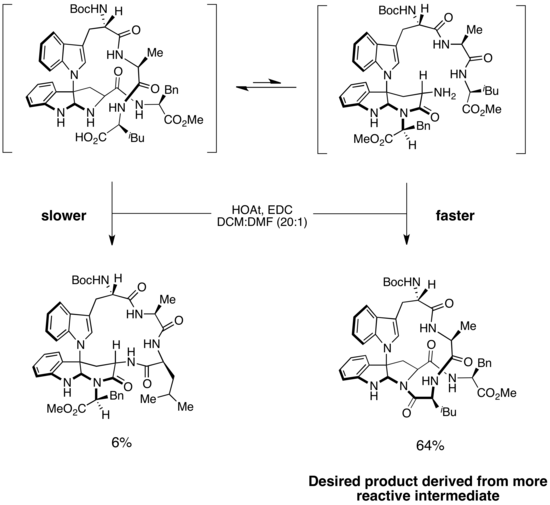
Síntesis de kapakahines B y F
Se invocó un escenario de Curtin-Hammett para explicar la selectividad en la síntesis de kapakahines B y F, dos péptidos cíclicos aislados de esponjas marinas. La estructura de cada uno de los dos compuestos contiene un macrociclo torcido de 16 miembros. Un paso clave en la síntesis es la formación selectiva de enlaces amida para producir el macrociclo correcto. En la síntesis enantioselectiva de las kapakahines B y F de Phil Baran, se propuso que la formación del macrociclo se produjera a través de dos isómeros del sustrato.El isómero de menor energía y más fácilmente accesible conducía al producto no deseado, mientras que el isómero menos estable formaba el producto deseado. Sin embargo, debido a que el paso de formación del enlace amida era irreversible y la barrera a la isomerización era baja, el producto principal se derivó del intermedio de reacción más rápida. Este es un ejemplo de un escenario de Curtin-Hammett en el que el intermedio menos estable es significativamente más reactivo que el intermedio más estable que predomina en la solución. Debido a que la isomerización del sustrato es rápida, durante el transcurso de la reacción, el exceso de sustrato de la forma más estable se puede convertir en la forma menos estable, que luego sufre una formación rápida e irreversible de enlaces amida para producir el macrociclo deseado. Esta estrategia proporcionó el producto deseado con una selectividad >10:1.(Creo que hay un error en el esquema. Consulte las páginas de discusión).
Síntesis de (+)-griseofulvina
En la primera síntesis enantioselectiva de (+)-Griseofulvina, un potente agente antifúngico, se observó una situación de Curtin-Hammett. Un paso clave en la síntesis es la formación catalizada por rodio de un iluro de oxonio, que luego sufre un reordenamiento sigmatrópico [2,3] en el camino hacia el producto deseado. Sin embargo, el sustrato contiene dos grupos orto-alcoxi, cualquiera de los cuales podría participar presumiblemente en la generación de iluro de oxonio.
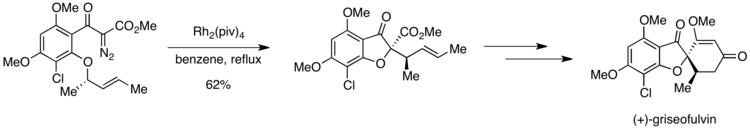
Sin embargo, fue posible obtener una alta selectividad para el producto deseado debido a las diferencias en las barreras de activación para el paso siguiente a la formación de iluro. Si el grupo orto-metoxi experimenta la formación de iluro de oxonio, un cambio de 1,4-metilo puede generar un producto no deseado. El iluro de oxonio formado a partir del otro grupo ortoalcoxi se ceba para sufrir una transposición sigmatrópica [2,3] para producir el compuesto deseado. Pirrung y colaboradores informaron una selectividad completa para el producto deseado sobre el producto resultante de un cambio de 1,4-metilo. Este resultado sugiere que la formación de iluro de oxonio es reversible, pero que el paso posterior es irreversible. El reordenamiento sigmatrópico [2,3] permitido por la simetría debe seguir una ruta que tiene una energía de activación más baja que el cambio de 1,4-metilo, lo que explica la formación exclusiva del producto deseado.

Síntesis de (+)-alociatina B 2
También se encontró un escenario potencial de Curtin-Hammett durante la síntesis total enantioselectiva de (+)-alociatina B2 por parte del grupo Trost.El paso fundamental en la síntesis fue una cicloisomerización diastereoselectiva catalizada por Ru. La reacción podría resultar en la formación de dos posibles isómeros de doble enlace. La reacción proporcionó una buena selectividad para el isómero deseado, con resultados consistentes con un escenario de Curtin-Hammett. La ciclorutenación oxidativa inicial y la eliminación de beta-hidruro producen un hidruro de vinil-rutenio. La inserción de hidruro permite una fácil isomerización de alquenos. Es poco probable que el resultado de la reacción refleje la estabilidad de los intermedios, ya que el gran grupo CpRu experimenta interacciones estéricas desfavorables con el grupo isopropilo cercano. En cambio, se aplica una situación de Curtin-Hammett, en la que el isómero favorecido en equilibrio no conduce al producto principal. La eliminación reductiva se ve favorecida por el intermedio más reactivo y menos estable, ya que el alivio de tensión se maximiza en el estado de transición. Esto produce el isómero de doble enlace deseado.

Contenido relacionado
Hidrógeno
Estereoquímica
Colágeno