
Isomería conformacional
En química, la isomería conformacional es una forma de estereoisomerismo en la que los isómeros pueden interconvertirse simplemente mediante rotaciones sobre enlaces formalmente simples (consulte la figura sobre la rotación de enlaces simples). Mientras que dos arreglos cualesquiera de átomos en una molécula que difieren por la rotación sobre enlaces sencillos pueden denominarse conformaciones diferentes, las conformaciones que corresponden a mínimos locales en la superficie de energía potencial se denominan específicamente isómeros o conformadores conformacionales.Las conformaciones que corresponden a máximos locales en la superficie de energía son los estados de transición entre los isómeros conformacionales mínimos locales. Las rotaciones sobre enlaces simples implican superar una barrera de energía rotacional para interconvertir un confórmero en otro. Si la barrera de energía es baja, hay rotación libre y existe una muestra del compuesto como una mezcla de múltiples confórmeros que se equilibra rápidamente; si la barrera de energía es lo suficientemente alta, entonces hay una rotación restringida, una molécula puede existir durante un período de tiempo relativamente largo como un isómero rotacional estable o rotámero(un isómero que surge de la rotación de un enlace simple impedida). Cuando la escala de tiempo para la interconversión es lo suficientemente larga para el aislamiento de rotámeros individuales (generalmente definido arbitrariamente como una vida media de interconversión de 1000 segundos o más), los isómeros se denominan atropisómeros (ver: atropisomerismo). El cambio de anillo de ciclohexanos sustituidos constituye otra forma común de isomería conformacional.
Los isómeros conformacionales son, por tanto, distintos de las otras clases de estereoisómeros (es decir, isómeros configuracionales) en los que la interconversión implica necesariamente la ruptura y reforma de enlaces químicos. Por ejemplo, las configuraciones L / D - y R / S - de moléculas orgánicas tienen diferentes actividades ópticas y manuales, y solo pueden interconvertirse rompiendo uno o más enlaces conectados al átomo quiral y reformando un enlace similar en una dirección diferente o espacial. orientación. También se diferencian de los geométricos (cis / trans) isómeros, otra clase de estereoisómeros, que requieren que el componente π de los dobles enlaces se rompa para la interconversión. (Aunque la distinción no siempre es clara, ya que ciertos enlaces que formalmente son enlaces simples en realidad tienen un carácter de enlace doble que se hace evidente solo cuando se consideran los contribuyentes de resonancia secundarios, como los enlaces C-N de las amidas, por ejemplo). Debido a interconversión rápida, los confórmeros no suelen ser aislables a temperatura ambiente.
El estudio de la energía entre diferentes conformaciones se conoce como análisis conformacional. Es útil para comprender la estabilidad de diferentes isómeros, por ejemplo, teniendo en cuenta la orientación espacial y las interacciones a través del espacio de los sustituyentes. Además, el análisis conformacional se puede utilizar para predecir y explicar la selectividad del producto, los mecanismos y las velocidades de las reacciones. El análisis conformacional también juega un papel importante en el diseño racional de fármacos basado en la estructura.
Tipos
Al rotar sus enlaces carbono-carbono, las moléculas de etano y propano tienen tres mínimos de energía locales. Son estructural y energéticamente equivalentes, y se denominan confórmeros escalonados. Para cada molécula, los tres sustituyentes que emanan de cada enlace carbono-carbono están escalonados, con cada ángulo diedro H–C–C–H (y ángulo diedro H–C–C–CH 3 en el caso del propano) igual a 60° (o aproximadamente igual a 60° en el caso del propano). Las tres conformaciones eclipsadas, en las que los ángulos diedros son cero, son estados de transición (máximos de energía) que conectan dos mínimos de energía equivalentes, los confórmeros escalonados.
La molécula de butano es la molécula más simple para la cual las rotaciones de enlaces simples dan como resultado dos tipos de estructuras no equivalentes, conocidas como conformadores anti y torpe (ver figura).
Por ejemplo, el butano tiene tres confórmeros relacionados con sus dos grupos metilo (CH 3): dos confórmeros torpes, que tienen los metilos separados ±60° y son enantioméricos, y un confórmero anti, donde los cuatro centros de carbono son coplanares y los sustituyentes son Separados 180° (ver diagrama de energía libre de butano). La diferencia de energía entre gauche y anti es de 0,9 kcal/mol asociada con la energía de deformación del confórmero gauche. El anticonfórmero es, por tanto, el más estable (≈ 0 kcal/mol). Las tres conformaciones eclipsadas con ángulos diédricos de 0°, 120° y 240° son estados de transición entre confórmeros.Tenga en cuenta que las dos conformaciones eclipsadas tienen energías diferentes: a 0°, los dos grupos metilo se eclipsan, lo que da como resultado una energía mayor (≈ 5 kcal/mol) que a 120°, donde los grupos metilo se eclipsan con hidrógenos (≈ 3,5 kcal/mol).).
Si bien las moléculas simples pueden describirse mediante este tipo de conformaciones, las moléculas más complejas requieren el uso del sistema Klyne-Prelog para describir los diferentes conformadores.
En otros lugares se detallan ejemplos más específicos de isomería conformacional:
- Conformación de anillo
- Conformaciones de ciclohexano, incluyendo conformaciones de silla y de bote entre otras.
- Conformaciones de cicloalcanos, incluidos anillos medianos y macrociclos.
- Conformación de carbohidratos, que incluye conformaciones de ciclohexano, así como otros detalles.
- Tensión alílica: energética relacionada con la rotación sobre el enlace simple entre un carbono sp y un carbono sp.
- Atropisomerismo: debido a la rotación restringida sobre un enlace.
- Plegamiento, incluyendo la estructura secundaria y terciaria de biopolímeros (ácidos nucleicos y proteínas).
- Akamptisomerismo: debido a la inversión restringida de un ángulo de enlace.
Energía libre y equilibrios de isómeros conformacionales
Equilibrio de confórmeros
Los isómeros conformacionales existen en un equilibrio dinámico, donde las energías libres relativas de los isómeros determinan la población de cada isómero y la barrera de energía de rotación determina la tasa de interconversión entre los isómeros:

donde K es la constante de equilibrio, Δ G° es la diferencia de energía libre estándar entre los dos confórmeros en kcal/mol, R es la constante universal de los gases (1,987 × 10 kcal/mol K) y T es la temperatura del sistema en grados Kelvin. En unidades de kcal/mol a 298 K,

Así, cada 1,36 kcal/mol corresponde a un factor de aproximadamente 10 en términos de constante de equilibrio a temperaturas cercanas a la temperatura ambiente. (La " regla de 1.36 " es útil en general para la estimación de constantes de equilibrio a temperatura ambiente a partir de diferencias de energía libre. A temperaturas más bajas, se necesita una diferencia de energía más pequeña para obtener una constante de equilibrio dada).
Se dan tres isotermas en el diagrama que representa la distribución de equilibrio de dos confórmeros a diferentes temperaturas. A una diferencia de energía libre de 0 kcal/mol, esto da una constante de equilibrio de 1, lo que significa que existen dos confórmeros en una proporción de 1:1. Los dos tienen la misma energía libre; ninguno es más estable, por lo que ninguno predomina en comparación con el otro. Una diferencia negativa en la energía libre significa que un confórmero se interconvierte a una conformación termodinámicamente más estable, por lo que la constante de equilibrio siempre será mayor que 1. Por ejemplo, el Δ G° para la transformación del butano del confórmero torpe al confórmero anti es −0,47 kcal/mol a 298 K. Esto da una constante de equilibrio de aproximadamente 2,2 a favor de laanti confórmero, o una mezcla 31:69 de gauche: anti confórmeros en equilibrio. Por el contrario, una diferencia positiva en la energía libre significa que el confórmero ya es el más estable, por lo que la interconversión es un equilibrio desfavorable (K < 1). Incluso para cambios altamente desfavorables (Δ G° positivo grande), la constante de equilibrio entre dos confórmeros puede incrementarse aumentando la temperatura, de modo que la cantidad del confórmero menos estable presente en el equilibrio aumenta (aunque siempre permanece como el confórmero menor).
Distribución de la población de confórmeros
La distribución fraccional de la población de diferentes confórmeros sigue una distribución de Boltzmann:

El lado izquierdo es la proporción de confórmero i en una mezcla de equilibrio de M confórmeros en equilibrio termodinámico. En el lado derecho, E k (k = 1, 2,..., M) es la energía del confórmero k, R es la constante de gas ideal molar (aproximadamente igual a 8.314 J/(mol·K) o 1.987 cal/ (mol·K)), y T es la temperatura absoluta. El denominador del lado derecho es la función de partición.
Factores que contribuyen a la energía libre de los confórmeros
Los efectos de las interacciones electrostáticas y estéricas de los sustituyentes, así como las interacciones orbitales, como la hiperconjugación, son responsables de la relativa estabilidad de los confórmeros y sus estados de transición. Las contribuciones de estos factores varían según la naturaleza de los sustituyentes y pueden contribuir positiva o negativamente a la barrera energética. Los estudios computacionales de moléculas pequeñas como el etano sugieren que los efectos electrostáticos son los que más contribuyen a la barrera energética; sin embargo, la barrera se atribuye tradicionalmente principalmente a las interacciones estéricas.
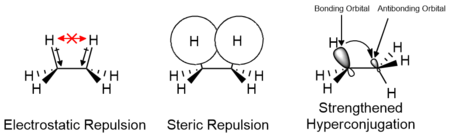 Contribuciones a la barrera de energía rotacional
Contribuciones a la barrera de energía rotacional
En el caso de sistemas cíclicos, el efecto estérico y la contribución a la energía libre se pueden aproximar mediante valores A, que miden la diferencia de energía cuando un sustituyente en el ciclohexano está en la posición axial en comparación con la ecuatorial. En anillos grandes (>14 átomos), hay muchas conformaciones accesibles de baja energía que corresponden a la red de diamante libre de tensión.
Aislamiento u observación de los isómeros conformacionales
La breve escala de tiempo de la interconversión impide la separación de los isómeros conformacionales en la mayoría de los casos. Los atropisómeros son isómeros conformacionales que se pueden separar debido a la rotación restringida. El equilibrio entre los isómeros conformacionales se puede observar usando una variedad de técnicas espectroscópicas.
El plegamiento de proteínas también genera isómeros conformacionales estables que se pueden observar. La ecuación de Karplus relaciona el ángulo diedro de los protones vecinales con sus constantes de acoplamiento J medidas por RMN. La ecuación ayuda a dilucidar el plegamiento de proteínas, así como las conformaciones de otras moléculas alifáticas rígidas. Las cadenas laterales de proteínas exhiben rotámeros, cuya distribución está determinada por su interacción estérica con diferentes conformaciones de la columna vertebral. Esto es evidente a partir del análisis estadístico de las conformaciones de las cadenas laterales de proteínas en la biblioteca de rotámeros dependientes de la columna vertebral.
En los derivados del ciclohexano, los dos confórmeros de silla se interconvierten rápidamente a temperatura ambiente, y el propio ciclohexano experimenta el cambio de anillo a una velocidad de aproximadamente 10 cambios de anillo/seg, con una barrera de energía total de 10 kcal/mol (42 kJ/mol).), lo que impide su separación a temperatura ambiente. Sin embargo, a bajas temperaturas por debajo del punto de coalescencia, se puede controlar directamente el equilibrio mediante espectroscopia de RMN y mediante espectroscopia de RMN dinámica, dependiente de la temperatura, la interconversión de barrera.
La dinámica de la isomería conformacional (y de otros tipos) se puede controlar mediante espectroscopia de RMN a temperaturas variables. La técnica se aplica a barreras de 8 a 14 kcal / mol, y las especies que exhiben tal dinámica a menudo se denominan "fluxionales".
Además de la espectroscopia de RMN, la espectroscopia de IR se utiliza para medir las proporciones de confórmeros. Para el confórmero axial y ecuatorial del bromociclohexano, ν CBr difiere en casi 50 cm.
Reacciones dependientes de la conformación
Las velocidades de reacción dependen en gran medida de la conformación de los reactivos. En muchos casos, el producto dominante surge de la reacción del conformador menos predominante, en virtud del principio de Curtin-Hammett. Esto es típico de situaciones en las que el equilibrio conformacional es mucho más rápido que la reacción para formar el producto. Por lo tanto, la dependencia de una reacción de la orientación estereoquímica generalmente solo es visible en los isómeros configuracionales, en los que los sustituyentes bloquean una conformación particular. La predicción de las tasas de muchas reacciones que implican la transición entre los estados sp2 y sp3, como la reducción de cetonas, la oxidación de alcoholes o la sustitución nucleófila, es posible si se tienen en cuenta todos los confórmeros y su estabilidad relativa regida por su cepa.
Un ejemplo con isómeros configuracionales lo proporcionan las reacciones de eliminación, que implican la eliminación simultánea de un protón y un grupo saliente de posiciones vecinales o antiperiplanares bajo la influencia de una base.
 Deshidrohalogenación bimolecular inducida por base (un mecanismo de reacción de tipo E2). La geometría óptima para el estado de transición requiere que los enlaces de ruptura sean antiperiplanares, ya que están en la conformación escalonada apropiada.
Deshidrohalogenación bimolecular inducida por base (un mecanismo de reacción de tipo E2). La geometría óptima para el estado de transición requiere que los enlaces de ruptura sean antiperiplanares, ya que están en la conformación escalonada apropiada.
El mecanismo requiere que los átomos o grupos que parten sigan trayectorias antiparalelas. Para sustratos de cadena abierta, este prerrequisito geométrico lo cumple al menos uno de los tres confórmeros escalonados. Sin embargo, para algunos sustratos cíclicos como el ciclohexano, es posible que no se pueda lograr una disposición antiparalela dependiendo de los sustituyentes que podrían establecer un bloqueo conformacional. Los sustituyentes adyacentes en un anillo de ciclohexano pueden lograr la antiperiplanaridad solo cuando ocupan posiciones transdiaxiales (es decir, ambos están en posición axial, uno hacia arriba y otro hacia abajo).
Una consecuencia de este análisis es que el cloruro de trans - 4 - terc -butilciclohexilo no puede eliminarse fácilmente, sino que sufre sustitución (ver diagrama a continuación) porque la conformación más estable tiene el voluminoso grupo t -Bu en la posición ecuatorial, por lo tanto, el grupo cloruro no es antiperiplanar con cualquier hidrógeno vecinal (es torpe para los cuatro). La conformación termodinámicamente desfavorecida tiene el grupo t - Bu en la posición axial, que tiene una energía más alta en más de 5 kcal/mol (ver valor A). Como resultado, el grupo t -Bu "bloquea" el anillo en la conformación donde está en la posición ecuatorial y se observa la reacción de sustitución. Por otro lado, cis -4-El cloruro de terc -butilciclohexilo se elimina porque se puede lograr la antiperiplanaridad de Cl y H cuando el grupo t -Bu está en la posición ecuatorial favorable.
Conformación termodinámicamente desfavorecida del cloruro de
trans - 4 -
terc -butilciclohexilo donde el grupo
t -Bu está en la posición axial ejerciendo interacciones de 7 átomos.

El isómero
trans puede alcanzar la antiperiplanaridad solo a través del confórmero axial desfavorecido; por lo tanto, no elimina. El isómero
cis ya tiene la geometría correcta en su conformación más estable; por lo tanto, se elimina fácilmente.
La repulsión entre un grupo t -butilo axial y los átomos de hidrógeno en la posición 1,3-diaxial es tan fuerte que el anillo de ciclohexano revertirá a una conformación de bote torcido. La deformación en las estructuras cíclicas suele caracterizarse por desviaciones de los ángulos de enlace ideales (deformación de Baeyer), ángulos de torsión ideales (deformación de Pitzer) o interacciones transanulares (Prelog).
Estereoquímica de alcanos
Los confórmeros de alcano surgen de la rotación alrededor de enlaces sigma carbono-carbono hibridados sp. El alcano más pequeño con tal enlace químico, el etano, existe como un número infinito de conformaciones con respecto a la rotación alrededor del enlace C-C. Dos de estos se reconocen como formas de energía mínima (conformación escalonada) y energía máxima (conformación eclipsada). La existencia de conformaciones específicas se debe a la rotación obstaculizada alrededor de los enlaces sigma, aunque una teoría competidora propone un papel para la hiperconjugación.
La importancia de los mínimos de energía y los máximos de energía se ve por la extensión de estos conceptos a moléculas más complejas para las cuales se pueden predecir conformaciones estables como formas de energía mínima. La determinación de conformaciones estables también ha jugado un papel importante en el establecimiento del concepto de inducción asimétrica y la capacidad de predecir la estereoquímica de reacciones controladas por efectos estéricos.
En el ejemplo del etano escalonado en la proyección de Newman, un átomo de hidrógeno en un átomo de carbono tiene un ángulo de torsión de 60° con respecto al átomo de hidrógeno más cercano en el otro carbono, de modo que se minimiza el impedimento estérico. La conformación escalonada es más estable en 12,5 kJ/mol que la conformación eclipsada, que es el máximo de energía para el etano. En la conformación eclipsada se minimiza el ángulo de torsión.



En el butano, las dos conformaciones escalonadas ya no son equivalentes y representan dos conformadores distintos: la anticonformación (más a la izquierda, abajo) y la conformación gauche (más a la derecha, abajo).




Ambas conformaciones están libres de tensión torsional, pero, en la conformación gauche, los dos grupos metilo están más cerca que la suma de sus radios de van der Waals. La interacción entre los dos grupos metilo es repulsiva (tensión de van der Waals) y se produce una barrera de energía.
Estos valores dan una medida de la energía potencial almacenada en los confórmeros de butano con mayor impedimento estérico que el estado fundamental del 'anti'conformador:
- Gauche, confórmero – 3,8 kJ/mol
- H y CH 3 eclipsados – 16 kJ/mol
- CH 3 y CH 3 eclipsados – 19 kJ/mol.
Los grupos metilo eclipsados ejercen una mayor tensión estérica debido a su mayor densidad de electrones en comparación con los átomos de hidrógeno solitarios.
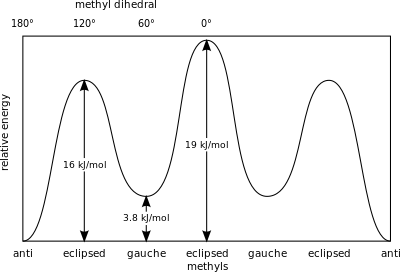 Energías relativas de las conformaciones del butano con respecto a la rotación del enlace CC central.
Energías relativas de las conformaciones del butano con respecto a la rotación del enlace CC central.
La explicación de libro de texto para la existencia del máximo de energía para una conformación eclipsada en el etano es el impedimento estérico, pero, con una longitud de enlace CC de 154 pm y un radio de Van der Waals para el hidrógeno de 120 pm, los átomos de hidrógeno en el etano nunca están en el camino del otro. La cuestión de si el impedimento estérico es responsable del máximo de energía eclipsado es un tema de debate hasta el día de hoy. Una alternativa a la explicación del impedimento estérico se basa en la hiperconjugación analizada dentro del marco del orbital de enlace natural.En la conformación escalonada, un orbital enlazante CH sigma dona densidad electrónica al orbital antienlazante del otro enlace CH. La estabilización energética de este efecto se maximiza cuando los dos orbitales se superponen al máximo, lo que ocurre en la conformación escalonada. No hay superposición en la conformación eclipsada, lo que lleva a un máximo de energía desfavorable. Por otro lado, un análisis dentro de la teoría cuantitativa de orbitales moleculares muestra que las repulsiones (estéricas) de 2 orbitales y 4 electrones son dominantes sobre la hiperconjugación. Un estudio de la teoría del enlace de valencia también enfatiza la importancia de los efectos estéricos.
Nomenclatura
La denominación de los alcanos según los estándares enumerados en el Libro de oro de la IUPAC se realiza de acuerdo con el sistema Klyne-Prelog para especificar ángulos (llamados ángulos de torsión o diedros) entre los sustituyentes alrededor de un enlace simple:

- un ángulo de torsión entre 0° y ± 90° se llama sin (s)
- un ángulo de torsión entre ± 90° y 180° se llama anti (a)
- un ángulo de torsión entre 30° y 150° o entre –30° y –150° se denomina clinal (c)
- un ángulo de torsión entre 0° y ± 30° o ± 150° y 180° se denomina periplanar (p)
- un ángulo de torsión entre 0° y ± 30° se llama sinperiplanar (sp), también llamado sin- o cis- conformación
- un ángulo de torsión entre 30° a 90° y –30° a –90° se llama sinclinal (sc), también llamado gauche o sesgado
- un ángulo de torsión entre 90° y 150° o –90° y –150° se llama anticlinal (ac)
- un ángulo de torsión entre ± 150° y 180° se llama antiperiplanar (ap), también llamado anti- o trans- conformación
La deformación por torsión o "deformación de Pitzer" se refiere a la resistencia a la torsión sobre un enlace.
Casos especiales
En el n -pentano, los grupos metilo terminales experimentan una interferencia adicional con el pentano.
Reemplazar el hidrógeno por flúor en el politetrafluoroetileno cambia la estereoquímica de la geometría en zigzag a la de una hélice debido a la repulsión electrostática de los átomos de flúor en las posiciones 1,3. La evidencia de la estructura de hélice en el estado cristalino se deriva de la cristalografía de rayos X y de la espectroscopia de RMN y el dicroísmo circular en solución.
Contenido relacionado
Ión poliatómico
Metaloide
Reglas de prioridad de Cahn-Ingold-Prelog