
Estereoselectividad
En química, la estereoselectividad es la propiedad de una reacción química en la que un solo reactivo forma una mezcla desigual de estereoisómeros durante una creación no estereoespecífica de un nuevo estereocentro o durante una transformación no estereoespecífica de uno preexistente. La selectividad surge de las diferencias en los efectos estéricos y electrónicos en las vías mecánicas que conducen a los diferentes productos. La estereoselectividad puede variar en grado pero nunca puede ser total ya que la diferencia de energía de activación entre las dos vías es finita. Ambos productos son al menos posibles y sólo difieren en la cantidad. Sin embargo, en casos favorables, el estereoisómero menor puede no ser detectable por los métodos analíticos utilizados.
Una reacción enantioselectiva es aquella en la que se forma un enantiómero con preferencia al otro, en una reacción que crea un producto ópticamente activo a partir de un material de partida aquiral, utilizando un catalizador quiral, una enzima o un reactivo quiral. El grado de selectividad se mide por el exceso enantiomérico. Una variante importante es la resolución cinética, en la que un centro quiral preexistente reacciona con un catalizador quiral, una enzima o un reactivo quiral de manera que un enantiómero reacciona más rápido que el otro y deja atrás el enantiómero menos reactivo, o en la que un enantiómero pre -el centro quiral existente influye en la reactividad de un centro de reacción en otra parte de la misma molécula.
Una reacción diastereoselectiva es aquella en la que se forma un diastereómero con preferencia a otro (o en la que un subconjunto de todos los diastereómeros posibles domina la mezcla de productos), estableciendo una estereoquímica relativa preferida. En este caso, se forman dos o más centros quirales a la vez de modo que se favorece una estereoquímica relativa, o un centro quiral preexistente (que no necesita ser ópticamente puro) sesga el resultado estereoquímico durante la creación de otro. El grado de selectividad relativa se mide por el exceso diastereoisómero.
La estereoconvergencia se puede considerar lo opuesto a la estereoespecificidad, cuando la reacción de dos estereoisómeros diferentes produce un solo producto estereoisómero.
La calidad de la estereoselectividad se refiere únicamente a los productos y su estereoquímica. De un número de posibles productos estereoisoméricos, la reacción selecciona uno o dos para que se formen.
Ejemplos
Un ejemplo de estereoselectividad modesta es la deshidrohalogenación de 2-yodo-butano que produce un 60% de trans -2-buteno y un 20% de cis -2-buteno. Dado que los isómeros geométricos de alquenos también se clasifican como diastereómeros, esta reacción también se denominaría diastereoselectiva.
La regla de Cram predice el diastereoisómero principal resultante de la adición nucleofílica diastereoselectiva a un grupo carbonilo próximo a un centro quiral. El centro quiral no necesita ser ópticamente puro, ya que la estereoquímica relativa será la misma para ambos enantiómeros. En el siguiente ejemplo, el (S)-aldehído reacciona con un tiazol para formar el diastereómero (S,S), pero solo una pequeña cantidad del diastereómero (S,R):
La epoxidación de Sharpless es un ejemplo de un proceso enantioselectivo, en el que un sustrato de alcohol alílico aquiral se transforma en un epoxialcohol ópticamente activo. En el caso de los alcoholes alílicos quirales, resulta la resolución cinética. Otro ejemplo es la dihidroxilación asimétrica de Sharpless. En el siguiente ejemplo, el alqueno aquiral produce solo uno de los 4 estereoisómeros posibles.
Con un centro estereogénico junto al carbocatión, la sustitución puede ser estereoselectiva en reacciones intermoleculares e intramoleculares. En la reacción que se muestra a continuación, el nucleófilo (furano) puede acercarse al carbocatión formado desde el lado menos protegido lejos del voluminoso grupo t-butilo, lo que da como resultado una alta diastereoselectividad facial:
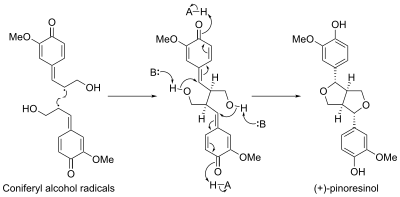
Biosíntesis estereoselectiva
La biosíntesis de pinoresinol involucró una proteína llamada proteína dirigente. La primera proteína dirigente fue descubierta en Forsythia intermedia. Se ha descubierto que esta proteína dirige la biosíntesis estereoselectiva de (+)-pinoresinol a partir de monómeros de alcohol coniferílico. Recientemente, se identificó una segunda proteína dirigente enantiocomplementaria en Arabidopsis thaliana, que dirige la síntesis enantioselectiva de (-)-pinoresinol.
Contenido relacionado
Ácido acetoacético
Pigmento de laca
Hierro