
Enfermedad renal poliquística autosómica dominante
La poliquistosis renal autosómica dominante (ADPKD) es uno de los trastornos humanos hereditarios más comunes y potencialmente mortales y la enfermedad renal hereditaria más común. Se asocia a una gran variabilidad interfamiliar e intrafamiliar, que puede explicarse en gran medida por su heterogeneidad genética y genes modificadores. También es la más común de las enfermedades renales quísticas hereditarias: un grupo de trastornos con una patogenia relacionada pero distinta, caracterizada por el desarrollo de quistes renales y diversas manifestaciones extrarrenales, que en el caso de la ADPKD incluyen quistes en otros órganos, como el hígado., vesículas seminales, páncreas y membrana aracnoidea, así como otras anomalías, como aneurismas y dolicoectasias intracraneales, dilatación y aneurismas de la raíz aórtica, prolapso de la válvula mitral y hernias de la pared abdominal. Más del 50% de los pacientes con ADPKD finalmente desarrollan enfermedad renal en etapa terminal y requieren diálisis o trasplante de riñón. Se estima que la ADPKD afecta al menos a una de cada 1000 personas en todo el mundo, lo que convierte a esta enfermedad en el trastorno renal hereditario más común con una prevalencia diagnosticada de 1:2000 y una incidencia de 1:3000-1:8000 a escala mundial.
Signos y síntomas
Entre las presentaciones clínicas se encuentran:
- Dolor agudo de lomo
- Sangre en la orina
- Riegos balotables
- Hemorragia subarachnoide (aeurisma de fresa)
- Hipertensión
- Cistos de hígado asociados
- Uremia por insuficiencia renal
- Anemia por enfermedad renal crónica
- Aumentar la secreción de RBC o eritropoyetina
Los signos y síntomas de ADPKD a menudo se desarrollan entre los 30 y los 40 años de edad.
Genética
La ADPKD es genéticamente heterogénea con dos genes identificados: PKD1 (región cromosómica 16p13.3; alrededor del 85 % de los casos) y PKD2 (4q21; alrededor del 15 % de los casos). Varios mecanismos genéticos probablemente contribuyen a la expresión fenotípica de la enfermedad. Aunque existe evidencia de un mecanismo de dos golpes (la línea germinal y la inactivación somática de dos alelos de PKD) que explican el desarrollo focal de quistes renales y hepáticos, es más probable que la haploinsuficiencia explique las manifestaciones vasculares de la enfermedad. Además, los nuevos modelos de ratones homocigotos para los alelos hipomórficos 22 y 23 de PKD1 y la demostración de una mayor proliferación de células epiteliales renales en ratones PKD2 +/− sugieren que otros mecanismos además de la hipótesis de los dos golpes también contribuyen al desarrollo quístico. fenotipo.
Ocurre una gran variabilidad interfamiliar e intrafamiliar en la ADPKD. La mayoría de las personas con mutaciones en PKD1 tienen insuficiencia renal a los 70 años, mientras que más del 50 % de las personas con mutaciones en PKD2 tienen una función renal adecuada a esa edad (edad media de aparición de enfermedad renal terminal: 54·3 años con PKD1; 74·0 años con PKD2).
La importante variabilidad intrafamiliar observada en la gravedad de las manifestaciones renales y extrarrenales apunta a factores modificadores genéticos y ambientales que pueden influir en el resultado de la ADPKD, y los resultados de un análisis de la variabilidad en la función renal entre gemelos monocigóticos y hermanos respaldan el papel de modificadores genéticos en esta enfermedad. Se estima que entre el 43% y el 78% de la variación en la edad de la ESRD podría deberse a factores modificadores hereditarios, siendo los padres tan propensos como los niños a mostrar una enfermedad más grave en estudios de parejas de padres e hijos.
Fisiopatología
En muchos pacientes con ADPKD, la disfunción renal no es clínicamente aparente hasta los 30 o 40 años de vida. Sin embargo, cada vez hay más evidencia que sugiere que la formación de quistes renales comienza en el útero. Los quistes se forman inicialmente como pequeñas dilataciones en los túbulos renales, que luego se expanden para formar cavidades llenas de líquido de diferentes tamaños. Los factores sugeridos para conducir a la cistogénesis incluyen una mutación de la línea germinal en uno de los alelos del gen de la policistina, un segundo golpe somático que conduce a la pérdida del alelo normal y un tercer golpe, que puede ser un daño renal que desencadena la proliferación celular, y un respuesta de lesión. Debido a numerosas similitudes entre la fisiopatología de la ADPKD y la fisiopatología de la respuesta renal a la lesión, la ADPKD se ha descrito como un estado de activación aberrante y persistente de las vías de respuesta a la lesión renal. En la progresión de la enfermedad, la dilatación continua de los túbulos a través del aumento de la proliferación celular, la secreción de líquido y la separación del túbulo parental conducen a la formación de quistes.
La PQRAD, junto con muchas otras enfermedades que cursan con quistes renales, se pueden clasificar en una familia de enfermedades conocidas como ciliopatías. Las células epiteliales de los túbulos renales, incluidos todos los segmentos de la nefrona y los conductos colectores (a excepción de las células intercaladas), muestran la presencia de un solo cilio apical primario. La policistina-1, la proteína codificada por el gen PKD1, está presente en estos cilios y se cree que detecta el flujo con sus grandes dominios extracelulares, activando los canales de calcio asociados con la policistina-2, el producto del gen PKD2, como resultado de el entorno genético de ADPKD como se explica en la subsección genética anterior.
La proliferación de células epiteliales y la secreción de fluidos que conducen a la cistogénesis son dos características distintivas de la ADPKD. Durante las primeras etapas de la cistogénesis, los quistes se adhieren a sus túbulos renales parentales y un derivado del filtrado glomerular ingresa a los quistes. Una vez que estos quistes se expanden a aproximadamente 2 mm de diámetro, el quiste se cierra de su túbulo parental y después de eso, el líquido solo puede ingresar a los quistes a través de la secreción transepitelial, que a su vez se sugiere que aumente debido a los efectos secundarios de una mayor concentración intracelular de cíclico. AMP (AMPc).
Desde el punto de vista clínico, el aumento insidioso del número y tamaño de los quistes renales se traduce en un aumento progresivo del volumen renal. Los estudios dirigidos por profesionales de Mayo Clinic establecieron que el volumen renal total (TKV) en una gran cohorte de pacientes con PQRAD fue de 1060 ± 642 ml con un aumento medio de 204 ml durante tres años, o 5,27 % por año en el curso natural de la enfermedad, entre otros hallazgos importantes y novedosos que se estudiaron extensamente por primera vez.

Diagnóstico
Por lo general, el diagnóstico de ADPKD se realiza inicialmente mediante imágenes renales mediante ultrasonido, tomografía computarizada o resonancia magnética. Sin embargo, el diagnóstico molecular puede ser necesario en las siguientes situaciones: 1- cuando se requiere un diagnóstico definitivo en personas jóvenes, como un posible donante vivo emparentado en una familia afectada con datos de imágenes equívocos; 2- en pacientes con antecedentes familiares negativos de ADPKD, debido a la posible superposición fenotípica con varias otras enfermedades quísticas renales; 3- en familias afectadas por poliquistosis renal de inicio precoz, ya que en estos casos pueden estar implicados alelos hipomórficos y/o herencia oligogénica; y 4- en pacientes que solicitan consejo genético, especialmente en parejas que desean un diagnóstico genético preimplantacional.
Los hallazgos de grandes riñones ecogénicos sin quistes macroscópicos distintivos en un bebé/niño con un riesgo del 50 % de ADPKD son diagnósticos. En ausencia de antecedentes familiares de ADPKD, la presencia de agrandamiento renal bilateral y quistes, con o sin la presencia de quistes hepáticos, y la ausencia de otras manifestaciones que sugieran una enfermedad quística renal diferente proporcionan evidencia presuntiva, pero no definitiva, de el diagnostico. En algunos casos, los aneurismas intracraneales pueden ser un signo asociado de ADPKD, y se puede recomendar la detección para pacientes con antecedentes familiares de aneurisma intracraneal.
Las pruebas genéticas moleculares mediante análisis de ligamiento o detección directa de mutaciones están clínicamente disponibles; sin embargo, la heterogeneidad genética es una complicación significativa para las pruebas genéticas moleculares. A veces, es necesario realizar pruebas a un número relativamente grande de miembros de la familia afectados para establecer cuál de los dos posibles genes es el responsable dentro de cada familia. El gran tamaño y la complejidad de los genes PKD1 y PKD2, así como la marcada heterogeneidad alélica, presentan obstáculos para las pruebas moleculares mediante análisis directo de ADN. La sensibilidad de la prueba es casi del 100% para todos los pacientes con ADPKD que tienen 30 años o más y para pacientes más jóvenes con mutaciones en PKD1; estos criterios solo tienen una sensibilidad del 67 % para los pacientes con mutaciones PKD2 menores de 30 años.

Riñón poliquístico adulto

Diagrama de enfermedad policítica dominante autosómica con inset renal normal para comparación
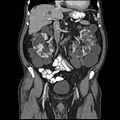
Tomografía computarizada abdominal de un adulto con enfermedad renal policítica dominante autosómica: La formación de quiste es extensa en ambos riñones, con algunos quistes en el hígado, también. (Avion coronal)



Tratamiento
Actualmente, el único tratamiento farmacológico disponible para la ADPKD consiste en reducir la velocidad de ganancia del volumen renal total (TKV) con antagonistas del receptor de vasopresina 2 (V2) (es decir, tolvaptán). El tratamiento con tolvaptán no detiene ni revierte la progresión de la enfermedad y los pacientes siguen progresando hacia la insuficiencia renal. Las modalidades de tratamiento paliativo incluyen medicamentos sintomáticos (analgésicos opioides y no opioides) para el dolor abdominal/retroperitoneal. Las opciones para el dolor resistente a los analgésicos incluyen procedimientos quirúrgicos simples o complejos (es decir, aspiración de quistes renales, decorticación de quistes, denervación renal y nefrectomía), que pueden provocar complicaciones inherentes a la cirugía. Investigaciones recientes sugieren que las intervenciones dietéticas cetogénicas afectan de manera beneficiosa la progresión y los síntomas en personas con ADPKD. La pérdida de peso leve afecta favorablemente el dolor, lo que indica el beneficio de los cambios en la dieta y el estilo de vida.
Medicación acuarética
En 2014, Japón fue el primer país del mundo en aprobar un tratamiento farmacológico para la PQRAD, seguido por Canadá y Europa, que aprobaron el fármaco tolvaptán para pacientes con PQRAD a principios de 2015. La FDA de EE. UU. aprobó el uso de tolvaptán en el tratamiento de la PQRAD en 2018. El tolvaptán, un fármaco acuarético, es un antagonista del receptor de vasopresina 2 (V2). Los estudios preclínicos sugirieron que la molécula cAMP podría estar involucrada en el agrandamiento de los quistes de ADPKD, y los estudios en roedores confirmaron el papel de la vasopresina en el aumento de los niveles de cAMP en el riñón, lo que sentó las bases para la realización de estudios clínicos. Debido a que los datos del Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) dirigido por Mayo Clinic mostraron que el volumen renal total (TKV) predijo el riesgo de desarrollar enfermedad renal crónica en pacientes con ADPKD, el ensayo TEMPO 3:4, que inscribió pacientes de 129 sitios en todo el mundo entre 2007 y 2009, evaluaron la TKV como criterio principal de valoración para probar la eficacia de tolvaptán en pacientes con ADPKD. Ese estudio mostró una disminución significativa en la proporción de aumento de TKV y disuasión de la disminución de la función renal en pacientes con ADPKD después del tratamiento con tolvaptán; sin embargo, debido a que los resultados de las pruebas de laboratorio con respecto a la función hepática parecían elevados en un porcentaje de pacientes inscritos en ese estudio, las agencias reguladoras retrasaron la aprobación del medicamento o, como en el caso de los EE. UU., la negaron por completo.
Intervenciones dietéticas y de estilo de vida
La investigación que utilizó modelos de ratones con ADPKD mostró que la restricción leve de alimentos mejoró considerablemente la progresión de la enfermedad. Se demostró que el mecanismo involucra el estado metabólico de la cetosis, y los efectos beneficiosos podrían producirse mediante la alimentación restringida en el tiempo, el ayuno agudo, una dieta cetogénica o mediante la suplementación con la cetona beta-hidroxibutirato en modelos de ADPKD en ratones, ratas y gatos. Un régimen de dieta cetogénica no solo detuvo la progresión de la enfermedad, sino que condujo a una reversión parcial de la enfermedad quística renal en un modelo de rata. El estado metabólico de la cetosis puede ser beneficioso en la ADPKD porque las células de los quistes renales en la ADPKD tienen un defecto metabólico similar al efecto Warburg en el cáncer que las hace altamente dependientes de la glucosa e incapaces de metabolizar los ácidos grasos y las cetonas. De acuerdo con esto, los niveles de glucosa sérica se correlacionan positivamente con una progresión más rápida de la enfermedad en pacientes con ADPKD. Además, las personas con ADPKD y diabetes tipo 2 tienen un volumen renal total (TKV) significativamente mayor que aquellos con ADPKD solo, y el sobrepeso o la obesidad se asocian con una progresión más rápida en la ADPKD en etapa temprana. Un estudio de serie de casos retrospectivo mostró que los síntomas de la enfermedad de PQRAD, incluidos el dolor, la hipertensión y la función renal, mejoraron entre 131 pacientes que implementaron dietas cetogénicas durante una duración promedio de 6 meses.
La ingesta de sodio en la dieta se asocia con una peor disminución de la función renal en pacientes con ADPKD y, en general, se recomienda limitar la ingesta de sodio a los pacientes. No se encontró que la ingesta de proteínas en la dieta se correlacionara con la progresión de la PQRAD.
Se cree que una mayor ingesta de agua es beneficiosa en la ADPKD y generalmente se recomienda. El mecanismo beneficioso subyacente del aumento de la ingesta de agua puede estar relacionado con los efectos sobre el receptor de vasopresina V2 o puede deberse a la supresión de la formación de microcristales nocivos en los túbulos renales mediante la dilución de solutos como el oxalato de calcio, el fosfato de calcio y el ácido úrico.
Se ha demostrado que la ingesta dietética de oxalato o fosfato inorgánico acelera la progresión de la enfermedad de PKD en varios modelos de roedores. Se ha demostrado que los niveles bajos de citrato urinario, un antagonista natural de la formación de cristales dañinos en los túbulos renales, se asocian con una peor progresión de la enfermedad en pacientes con ADPKD.
Medicación analgésica
El dolor crónico en pacientes con ADPKD a menudo es refractario a los tratamientos conservadores no invasivos, pero los analgésicos no opioides y las intervenciones conservadoras pueden usarse primero antes de considerar los analgésicos opioides; si el dolor continúa, las intervenciones quirúrgicas pueden dirigirse a los quistes renales o hepáticos para abordar directamente la causa del dolor, con opciones quirúrgicas que incluyen la decorticación del quiste renal, la denervación renal y la nefrectomía.
Aspiración de quiste renal
La aspiración con escleroterapia con etanol se puede realizar para el tratamiento de quistes renales simples sintomáticos, pero puede resultar poco práctica en pacientes avanzados con quistes múltiples. El procedimiento en sí consiste en la inserción percutánea de una aguja en el quiste identificado, bajo guía ecográfica, con posterior drenaje del líquido contenido; la escleroterapia se usa para evitar la reacumulación de líquido que puede ocurrir en el quiste, lo que puede resultar en la recurrencia de los síntomas.
Decorticación laparoscópica de quistes
La decorticación laparoscópica de quistes (también conocida como marsupialización) consiste en la extirpación de uno o más quistes renales mediante cirugía laparoscópica, durante la cual se perforan los quistes y se extirpa la pared exterior de los quistes más grandes con cuidado de no incidir el riñón. parénquima. Este procedimiento puede ser útil para aliviar el dolor en pacientes con ADPKD, y generalmente está indicado después de que la aspiración previa del quiste haya confirmado que el quiste que se va a descortezar es el responsable del dolor. Los ensayos controlados no aleatorizados realizados en la década de 1990 demostraron que los pacientes con quistes renales simples sintomáticos que presentaban recurrencia de los síntomas después de la respuesta inicial a la aspiración simple podían someterse de forma segura a la decorticación del quiste, con una vida media sin dolor de entre 17 y 24 meses. después de cirugía. La decorticación laparoscópica presenta una tasa de recurrencia de quistes renales del 5% en comparación con una tasa de recurrencia del 82% obtenida con la escleroterapia.
Neurólisis
Un tratamiento novedoso específicamente para el dolor crónico experimentado por muchas personas con ADPKD es la neurólisis del plexo celíaco. Esto implica la ablación química del plexo celíaco, para causar una degeneración temporal de las fibras nerviosas específicas. Cuando las fibras nerviosas se degeneran, se produce una interrupción en la transmisión de las señales nerviosas. Este tratamiento, cuando tiene éxito, proporciona un alivio significativo del dolor durante un período que va desde unos pocos días hasta más de un año. El procedimiento puede repetirse cuando los nervios afectados se hayan curado y el dolor regrese.
Nefrectomía
Muchos pacientes con ADPKD experimentan secuelas sintomáticas como consecuencia de la enfermedad, como hemorragia del quiste, dolor en el costado, infecciones recurrentes, nefrolitiasis y síntomas de efecto de masa (es decir, saciedad temprana, náuseas y vómitos y molestias abdominales), a partir de su riñones agrandados. En tales casos, puede ser necesaria la nefrectomía debido a síntomas intratables o cuando, en el curso de la preparación para el trasplante de riñón, se descubre que los riñones nativos comprimen la pelvis verdadera e impiden la colocación de un aloinjerto de donante. Además, la nefrectomía nativa se puede realizar en presencia de sospecha de malignidad, ya que el carcinoma de células renales (RCC) es de dos a tres veces más probable en la población con ADPKD en la enfermedad renal en etapa terminal (ESKD) que en los pacientes con ESKD sin ADPKD. Aunque las indicaciones para la nefrectomía en ADPKD pueden estar relacionadas con el tamaño del riñón, la decisión de proceder con la nefrectomía nativa a menudo se toma de forma individual, sin referencia específica a las medidas del tamaño del riñón.
Diálisis
Se pueden utilizar dos modalidades de diálisis en el tratamiento de pacientes con ADPKD: diálisis peritoneal y hemodiálisis. Los datos epidemiológicos muestran que la ADPKD afecta al 5-13,4 % de los pacientes sometidos a hemodiálisis en Europa y Estados Unidos, y alrededor del 3 % en Japón. La diálisis peritoneal generalmente ha estado contraindicada en pacientes con PQRAD con grandes volúmenes renales y hepáticos, debido a las dificultades físicas esperadas en el procedimiento y posibles complicaciones; sin embargo, no se observa ninguna diferencia en la morbilidad a largo plazo entre la hemodiálisis y la diálisis peritoneal en ADPKD.
Trasplante de riñón
El trasplante de riñón se acepta como el tratamiento preferido para pacientes con ADPKD con ESRD. Entre los pacientes estadounidenses en lista de espera para trasplante de riñón (a diciembre de 2011), 7256 (8,4 %) se incluyeron debido a enfermedad renal quística y de los 16 055 trasplantes renales realizados en 2011, 2057 (12,8 %) se realizaron para pacientes con enfermedad renal quística. enfermedad renal, con 1.189 de donante cadáver y 868 de donante vivo.
Pronóstico
En los pacientes con ADPKD, el desarrollo y la expansión graduales del quiste dan como resultado el agrandamiento del riñón y, durante el curso de la enfermedad, la tasa de filtración glomerular permanece normal durante décadas antes de que la función renal comience a deteriorarse progresivamente, lo que dificulta la predicción temprana del resultado renal. El estudio CRISP, mencionado en la sección anterior de tratamiento, contribuyó a construir una base sólida que apoya el valor pronóstico del volumen renal total (TKV) en ADPKD; El TKV (evaluado por resonancia magnética) aumenta constantemente y una mayor tasa de agrandamiento del riñón se correlaciona con una disminución acelerada de la TFG, mientras que el TKV ajustado a la altura del paciente (HtTKV) ≥600 ml/m predice el desarrollo de enfermedad renal crónica en etapa 3 dentro de los 8 años.
Además de TKV y HtTKV, la tasa de filtración glomerular estimada (eGFR) también se ha utilizado tentativamente para predecir la progresión de ADPKD. Después del análisis de tomografías computarizadas o resonancias magnéticas de 590 pacientes con ADPKD tratados en el Centro de enfermedad renal poliquística traslacional de Mayo, Irazabal y sus colegas desarrollaron un sistema de clasificación basado en imágenes para predecir la tasa de disminución de eGFR en pacientes con ADPKD. En este método de pronóstico, los pacientes se dividen en cinco subclases de tasas de crecimiento renal estimadas según los rangos de HtTKV específicos de la edad (1A, <1,5 %; 1B, 1,5–3,0 %; 1C, 3,0–4,5 %; 1D, 4,5–6,0 % y 1E, >6,0 %) como se indica en el estudio CRISP. La disminución de eGFR durante los años posteriores a la medición inicial de TKV es significativamente diferente entre las cinco subclases de pacientes, y los de la subclase 1E tienen la disminución más rápida. Algunas de las causas más comunes de muerte en pacientes con ADPKD son diversas infecciones (25 %), rotura de un aneurisma de bayas (15 %) o cardiopatía coronaria/hipertensiva (40 %).
Contenido relacionado
Lipoproteína
Antidepresivo tricíclico
Alois alzheimer