
Bioinformática
La bioinformática es un campo interdisciplinario que desarrolla métodos y herramientas de software para comprender datos biológicos, en particular cuando los conjuntos de datos son grandes y complejos.. Como campo interdisciplinario de la ciencia, la bioinformática combina la biología, la química, la física, la informática, la ingeniería de la información, las matemáticas y la estadística para analizar e interpretar los datos biológicos. La bioinformática se ha utilizado para in silicoanálisis de consultas biológicas utilizando técnicas computacionales y estadísticas.
La bioinformática incluye estudios biológicos que utilizan la programación informática como parte de su metodología, así como "tuberías" de análisis específicas que se utilizan repetidamente, en particular en el campo de la genómica. Los usos comunes de la bioinformática incluyen la identificación de genes candidatos y polimorfismos de un solo nucleótido (SNP). A menudo, dicha identificación se realiza con el objetivo de comprender mejor la base genética de la enfermedad, las adaptaciones únicas, las propiedades deseables (especialmente en especies agrícolas) o las diferencias entre poblaciones. De una manera menos formal, la bioinformática también trata de comprender los principios organizativos dentro de las secuencias de ácidos nucleicos y proteínas, lo que se denomina proteómica.
El procesamiento de imágenes y señales permite la extracción de resultados útiles a partir de grandes cantidades de datos sin procesar. En el campo de la genética, ayuda a secuenciar y anotar genomas y sus mutaciones observadas. Desempeña un papel en la extracción de textos de la literatura biológica y el desarrollo de ontologías biológicas y genéticas para organizar y consultar datos biológicos. También juega un papel en el análisis de la expresión y regulación de genes y proteínas. Las herramientas bioinformáticas ayudan a comparar, analizar e interpretar datos genéticos y genómicos y, de manera más general, a comprender los aspectos evolutivos de la biología molecular. En un nivel más integrador, ayuda a analizar y catalogar las rutas y redes biológicas que son una parte importante de la biología de sistemas. En biología estructural, ayuda en la simulación y modelado de ADN, ARN,proteínas e interacciones biomoleculares.
Historia
Históricamente, el término bioinformática no significaba lo que significa hoy. Paulien Hogeweg y Ben Hesper lo acuñaron en 1970 para referirse al estudio de los procesos de información en los sistemas bióticos. Esta definición colocó a la bioinformática como un campo paralelo a la bioquímica (el estudio de los procesos químicos en los sistemas biológicos).
Secuencias
Ha habido un tremendo avance en velocidad y reducción de costos desde la finalización del Proyecto Genoma Humano, con algunos laboratorios capaces de secuenciar más de 100,000 billones de bases cada año, y se puede secuenciar un genoma completo por mil dólares o menos. Las computadoras se volvieron esenciales en biología molecular cuando las secuencias de proteínas estuvieron disponibles después de que Frederick Sanger determinara la secuencia de la insulina a principios de la década de 1950. Comparar múltiples secuencias manualmente resultó ser poco práctico. Una pionera en el campo fue Margaret Oakley Dayhoff. Compiló una de las primeras bases de datos de secuencias de proteínas, publicada inicialmente como libros y fue pionera en métodos de alineación de secuencias y evolución molecular.Otro de los primeros contribuyentes a la bioinformática fue Elvin A. Kabat, quien fue pionero en el análisis de secuencias biológicas en 1970 con sus volúmenes completos de secuencias de anticuerpos publicados con Tai Te Wu entre 1980 y 1991. En la década de 1970, se aplicaron nuevas técnicas para secuenciar el ADN al bacteriófago MS2 y øX174, y las secuencias de nucleótidos extendidas luego se analizaron con algoritmos informativos y estadísticos. Estos estudios ilustraron que las características bien conocidas, como los segmentos de codificación y el código de triplete, se revelan en análisis estadísticos sencillos y, por lo tanto, fueron una prueba del concepto de que la bioinformática sería reveladora.
Metas
Para estudiar cómo se alteran las actividades celulares normales en diferentes estados de enfermedad, los datos biológicos deben combinarse para formar una imagen completa de estas actividades. Por lo tanto, el campo de la bioinformática ha evolucionado de tal manera que la tarea más apremiante implica ahora el análisis y la interpretación de varios tipos de datos. Esto también incluye secuencias de nucleótidos y aminoácidos, dominios de proteínas y estructuras de proteínas. El proceso real de análisis e interpretación de datos se conoce como biología computacional. Las subdisciplinas importantes dentro de la bioinformática y la biología computacional incluyen:
- Desarrollo e implementación de programas informáticos que permitan el acceso, manejo y uso eficiente de diversos tipos de información.
- Desarrollo de nuevos algoritmos (fórmulas matemáticas) y medidas estadísticas que evalúan las relaciones entre los miembros de grandes conjuntos de datos. Por ejemplo, existen métodos para localizar un gen dentro de una secuencia, para predecir la estructura y/o función de una proteína y para agrupar secuencias de proteínas en familias de secuencias relacionadas.
El objetivo principal de la bioinformática es aumentar la comprensión de los procesos biológicos. Sin embargo, lo que lo distingue de otros enfoques es su enfoque en el desarrollo y la aplicación de técnicas computacionalmente intensivas para lograr este objetivo. Los ejemplos incluyen: reconocimiento de patrones, minería de datos, algoritmos de aprendizaje automático y visualización. Los principales esfuerzos de investigación en el campo incluyen la alineación de secuencias, la búsqueda de genes, el ensamblaje del genoma, el diseño de fármacos, el descubrimiento de fármacos, la alineación de la estructura de las proteínas, la predicción de la estructura de las proteínas, la predicción de la expresión génica y las interacciones proteína-proteína, los estudios de asociación del genoma completo, el modelado de la evolución y división celular/mitosis.
La bioinformática ahora implica la creación y el avance de bases de datos, algoritmos, técnicas computacionales y estadísticas y teoría para resolver problemas formales y prácticos que surgen del manejo y análisis de datos biológicos.
Durante las últimas décadas, los rápidos desarrollos en genómica y otras tecnologías de investigación molecular y los avances en tecnologías de la información se han combinado para producir una gran cantidad de información relacionada con la biología molecular. Bioinformática es el nombre que se le da a estos enfoques matemáticos y computacionales que se utilizan para comprender mejor los procesos biológicos.
Las actividades comunes en bioinformática incluyen el mapeo y el análisis de secuencias de proteínas y ADN, la alineación de secuencias de proteínas y ADN para compararlas, y la creación y visualización de modelos tridimensionales de estructuras de proteínas.
Relación con otros campos
La bioinformática es un campo de la ciencia que es similar pero distinto de la computación biológica, mientras que a menudo se considera sinónimo de biología computacional. La computación biológica usa la bioingeniería y la biología para construir computadoras biológicas, mientras que la bioinformática usa la computación para comprender mejor la biología. La bioinformática y la biología computacional involucran el análisis de datos biológicos, particularmente secuencias de ADN, ARN y proteínas. El campo de la bioinformática experimentó un crecimiento explosivo a partir de mediados de la década de 1990, impulsado en gran parte por el Proyecto Genoma Humano y por los rápidos avances en la tecnología de secuenciación del ADN.
El análisis de datos biológicos para producir información significativa implica escribir y ejecutar programas de software que utilizan algoritmos de teoría de grafos, inteligencia artificial, informática, extracción de datos, procesamiento de imágenes y simulación por computadora. Los algoritmos a su vez dependen de fundamentos teóricos como las matemáticas discretas, la teoría de control, la teoría de sistemas, la teoría de la información y la estadística.
Análisis de secuencia
Desde que se secuenció el fago Φ-X174 en 1977, las secuencias de ADN de miles de organismos se han decodificado y almacenado en bases de datos. Esta información de secuencia se analiza para determinar genes que codifican proteínas, genes de ARN, secuencias reguladoras, motivos estructurales y secuencias repetitivas. Una comparación de genes dentro de una especie o entre diferentes especies puede mostrar similitudes entre funciones de proteínas o relaciones entre especies (el uso de la sistemática molecular para construir árboles filogenéticos). Con la creciente cantidad de datos, hace mucho tiempo que se volvió poco práctico analizar secuencias de ADN manualmente. Los programas informáticos como BLAST se utilizan habitualmente para buscar secuencias; a partir de 2008, de más de 260 000 organismos, que contienen más de 190 000 millones de nucleótidos.
Secuencia ADN
Antes de que las secuencias puedan analizarse, deben obtenerse del banco de almacenamiento de datos, por ejemplo, Genbank. La secuenciación del ADN sigue siendo un problema no trivial, ya que los datos sin procesar pueden ser ruidosos o verse afectados por señales débiles. Se han desarrollado algoritmos para llamar a la base de los diversos enfoques experimentales para la secuenciación del ADN.
Montaje de secuencia
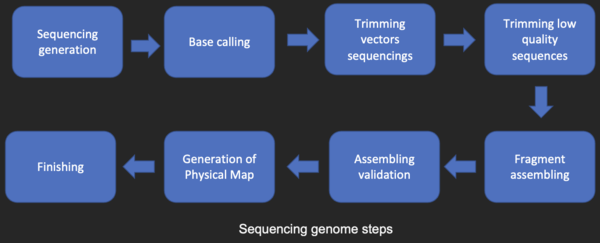
La mayoría de las técnicas de secuenciación de ADN producen fragmentos cortos de secuencia que deben ensamblarse para obtener secuencias completas de genes o genomas. La llamada técnica de secuenciación de escopeta (que fue utilizada, por ejemplo, por The Institute for Genomic Research (TIGR) para secuenciar el primer genoma bacteriano, Haemophilus influenzae)genera las secuencias de muchos miles de pequeños fragmentos de ADN (que van desde 35 a 900 nucleótidos de largo, dependiendo de la tecnología de secuenciación). Los extremos de estos fragmentos se superponen y, cuando se alinean correctamente mediante un programa de ensamblaje del genoma, se pueden utilizar para reconstruir el genoma completo. La secuenciación de escopeta produce datos de secuencia rápidamente, pero la tarea de ensamblar los fragmentos puede ser bastante complicada para genomas más grandes. Para un genoma tan grande como el genoma humano, puede tomar muchos días de tiempo de CPU en computadoras multiprocesador de gran memoria para ensamblar los fragmentos, y el ensamblaje resultante generalmente contiene numerosos vacíos que deben llenarse más tarde. La secuenciación de escopeta es el método de elección para prácticamente todos los genomas secuenciados en la actualidad, y los algoritmos de ensamblaje de genomas son un área crítica de la investigación bioinformática.
Anotación del genoma
En el contexto de la genómica, la anotación es el proceso de marcar los genes y otras características biológicas en una secuencia de ADN. Este proceso debe automatizarse porque la mayoría de los genomas son demasiado grandes para anotarlos a mano, sin mencionar el deseo de anotar tantos genomas como sea posible, ya que la velocidad de secuenciación ha dejado de representar un cuello de botella. La anotación es posible gracias al hecho de que los genes tienen regiones de inicio y finalización reconocibles, aunque la secuencia exacta que se encuentra en estas regiones puede variar entre genes.
La anotación del genoma se puede clasificar en tres niveles: los niveles de nucleótidos, proteínas y procesos.
El hallazgo de genes es un aspecto principal de la anotación a nivel de nucleótidos. Para genomas complejos, los métodos más exitosos utilizan una combinación de predicción de genes ab initio y comparación de secuencias con bases de datos de secuencias expresadas y otros organismos. La anotación a nivel de nucleótidos también permite la integración de la secuencia del genoma con otros mapas genéticos y físicos del genoma.
El objetivo principal de la anotación a nivel de proteína es asignar una función a los productos del genoma. Las bases de datos de secuencias de proteínas y dominios y motivos funcionales son recursos poderosos para este tipo de anotación. Sin embargo, la mitad de las proteínas predichas en una nueva secuencia del genoma tienden a no tener una función obvia.
Comprender la función de los genes y sus productos en el contexto de la fisiología celular y del organismo es el objetivo de la anotación a nivel de proceso. Uno de los obstáculos a este nivel de anotación ha sido la inconsistencia de los términos utilizados por diferentes sistemas modelo. El Gene Ontology Consortium está ayudando a resolver este problema.
La primera descripción de un sistema completo de anotación del genoma fue publicada en 1995 por el equipo del Instituto de Investigación Genómica que realizó la primera secuenciación y análisis completos del genoma de un organismo de vida libre, la bacteria Haemophilus influenzae. Owen White diseñó y construyó un sistema de software para identificar los genes que codifican todas las proteínas, transferir ARN, ARN ribosomal (y otros sitios) y realizar asignaciones funcionales iniciales. La mayoría de los sistemas actuales de anotación del genoma funcionan de manera similar, pero los programas disponibles para el análisis del ADN genómico, como el programa GeneMark entrenado y utilizado para encontrar genes que codifican proteínas en Haemophilus influenzae, cambian y mejoran constantemente.
Siguiendo las metas que el Proyecto Genoma Humano dejó de alcanzar tras su cierre en 2003, apareció un nuevo proyecto desarrollado por el Instituto Nacional de Investigación del Genoma Humano de EE.UU. El llamado proyecto ENCODE es una recopilación colaborativa de datos de los elementos funcionales del genoma humano que utiliza tecnologías de secuenciación de ADN de próxima generación y matrices de mosaico genómico, tecnologías capaces de generar automáticamente grandes cantidades de datos a un costo por base drásticamente reducido. pero con la misma precisión (error de llamada de base) y fidelidad (error de montaje).
Predicción de la función genética
Si bien la anotación del genoma se basa principalmente en la similitud de la secuencia (y, por lo tanto, en la homología), se pueden usar otras propiedades de las secuencias para predecir la función de los genes. De hecho, la mayoría de los métodos de predicción de la función de los genes se centran en las secuencias de proteínas, ya que son más informativas y ricas en características. Por ejemplo, la distribución de aminoácidos hidrofóbicos predice segmentos transmembrana en proteínas. Sin embargo, la predicción de la función de las proteínas también puede utilizar información externa, como datos de expresión de genes (o proteínas), estructura de proteínas o interacciones proteína-proteína.
Biología evolutiva computacional
La biología evolutiva es el estudio del origen y descendencia de las especies, así como su cambio a lo largo del tiempo. La informática ha ayudado a los biólogos evolutivos al permitirles a los investigadores:
- rastrear la evolución de una gran cantidad de organismos midiendo los cambios en su ADN, en lugar de solo a través de la taxonomía física o las observaciones fisiológicas,
- comparar genomas completos, lo que permite el estudio de eventos evolutivos más complejos, como la duplicación de genes, la transferencia horizontal de genes y la predicción de factores importantes en la especiación bacteriana,
- construir modelos computacionales complejos de genética de poblaciones para predecir el resultado del sistema a lo largo del tiempo
- rastrear y compartir información sobre un número cada vez mayor de especies y organismos
El trabajo futuro se esfuerza por reconstruir el ahora más complejo árbol de la vida.
El área de investigación dentro de la informática que utiliza algoritmos genéticos a veces se confunde con la biología evolutiva computacional, pero las dos áreas no están necesariamente relacionadas.
Genómica comparativa
El núcleo del análisis comparativo del genoma es el establecimiento de la correspondencia entre genes (análisis de ortología) u otras características genómicas en diferentes organismos. Son estos mapas intergenómicos los que permiten rastrear los procesos evolutivos responsables de la divergencia de dos genomas. Una multitud de eventos evolutivos que actúan en varios niveles organizacionales dan forma a la evolución del genoma. En el nivel más bajo, las mutaciones puntuales afectan a los nucleótidos individuales. En un nivel superior, los grandes segmentos cromosómicos sufren duplicación, transferencia lateral, inversión, transposición, eliminación e inserción.En última instancia, los genomas completos están involucrados en procesos de hibridación, poliploidización y endosimbiosis, que a menudo conducen a una rápida especiación. La complejidad de la evolución del genoma plantea muchos desafíos emocionantes para los desarrolladores de modelos matemáticos y algoritmos, que recurren a un espectro de técnicas algorítmicas, estadísticas y matemáticas, que van desde algoritmos exactos, heurísticos, de parámetros fijos y de aproximación para problemas basados en modelos de parsimonia hasta Markov. Algoritmos de Monte Carlo en cadena para el análisis bayesiano de problemas basados en modelos probabilísticos.
Muchos de estos estudios se basan en la detección de homología de secuencia para asignar secuencias a familias de proteínas.
Pangenómica
Pangenomics es un concepto introducido en 2005 por Tettelin y Medini que finalmente echó raíces en la bioinformática. El genoma pan es el repertorio completo de genes de un grupo taxonómico particular: aunque inicialmente se aplicó a cepas estrechamente relacionadas de una especie, se puede aplicar a un contexto más amplio como género, phylum, etc. Se divide en dos partes: el genoma central: Conjunto de genes comunes a todos los genomas en estudio (a menudo son genes domésticos vitales para la supervivencia) y el genoma prescindible/flexible: conjunto de genes que no están presentes en todos excepto en uno o algunos genomas en estudio. Se puede utilizar una herramienta bioinformática BPGA para caracterizar el Genoma Pan de especies bacterianas.
Genética de la enfermedad
Con el advenimiento de la secuenciación de próxima generación, estamos obteniendo suficientes datos de secuencia para mapear los genes de enfermedades complejas, como la infertilidad, el cáncer de mama o la enfermedad de Alzheimer. Los estudios de asociación del genoma completo son un enfoque útil para identificar las mutaciones responsables de enfermedades tan complejas. A través de estos estudios, se han identificado miles de variantes de ADN que están asociadas con enfermedades y rasgos similares. Además, la posibilidad de que los genes se utilicen en el pronóstico, diagnóstico o tratamiento es una de las aplicaciones más esenciales. Muchos estudios están discutiendo tanto las formas prometedoras de elegir los genes que se usarán como los problemas y peligros de usar genes para predecir la presencia o el pronóstico de una enfermedad.
Análisis de mutaciones en cáncer
En el cáncer, los genomas de las células afectadas se reorganizan de formas complejas o incluso impredecibles. Los esfuerzos de secuenciación masiva se utilizan para identificar mutaciones puntuales previamente desconocidas en una variedad de genes en el cáncer. Los bioinformáticos continúan produciendo sistemas automatizados especializados para administrar el gran volumen de datos de secuencia producidos, y crean nuevos algoritmos y software para comparar los resultados de secuenciación con la creciente colección de secuencias del genoma humano y polimorfismos de la línea germinal. Se emplean nuevas tecnologías de detección física, como micromatrices de oligonucleótidos para identificar ganancias y pérdidas cromosómicas (llamada hibridación genómica comparativa), y matrices de polimorfismos de un solo nucleótido para detectar mutaciones puntuales conocidas.. Estos métodos de detección miden simultáneamente varios cientos de miles de sitios en todo el genoma y, cuando se utilizan con un alto rendimiento para medir miles de muestras, generan terabytes de datos por experimento. Una vez más, las cantidades masivas y los nuevos tipos de datos generan nuevas oportunidades para los bioinformáticos. A menudo se encuentra que los datos contienen una variabilidad considerable, o ruido, y, por lo tanto, se están desarrollando métodos de análisis de punto de cambio y modelo oculto de Markov para inferir cambios reales en el número de copias.
Se pueden utilizar dos principios importantes en el análisis bioinformático de los genomas del cáncer relacionados con la identificación de mutaciones en el exoma. Primero, el cáncer es una enfermedad de mutaciones somáticas acumuladas en los genes. El segundo cáncer contiene mutaciones del conductor que deben distinguirse de los pasajeros.
Con los avances que esta tecnología de secuenciación de próxima generación está brindando al campo de la bioinformática, la genómica del cáncer podría cambiar drásticamente. Estos nuevos métodos y software permiten a los bioinformáticos secuenciar muchos genomas de cáncer de forma rápida y económica. Esto podría crear un proceso más flexible para clasificar los tipos de cáncer mediante el análisis de mutaciones en el genoma provocadas por el cáncer. Además, el seguimiento de pacientes mientras la enfermedad progresa puede ser posible en el futuro con la secuencia de muestras de cáncer.
Otro tipo de datos que requiere un desarrollo informático novedoso es el análisis de lesiones que se encuentran recurrentes entre muchos tumores.
Expresión de genes y proteínas
Análisis de la expresión génica
La expresión de muchos genes se puede determinar midiendo los niveles de ARNm con múltiples técnicas que incluyen micromatrices, secuenciación de etiquetas de secuencia de ADNc expresada (EST), secuenciación de etiquetas de análisis en serie de expresión génica (SAGE), secuenciación de firmas paralelas masivas (MPSS), RNA-Seq, también conocido como "Secuenciación de escopeta de transcriptoma completo" (WTSS), o varias aplicaciones de hibridación in situ multiplexada. Todas estas técnicas son extremadamente propensas al ruido y/o sujetas a sesgos en la medición biológica, y un área importante de investigación en biología computacional involucra el desarrollo de herramientas estadísticas para separar la señal del ruido en estudios de expresión génica de alto rendimiento.Dichos estudios a menudo se utilizan para determinar los genes implicados en un trastorno: uno podría comparar datos de micromatrices de células epiteliales cancerosas con datos de células no cancerosas para determinar las transcripciones que están reguladas al alza y a la baja en una población particular de células cancerosas..
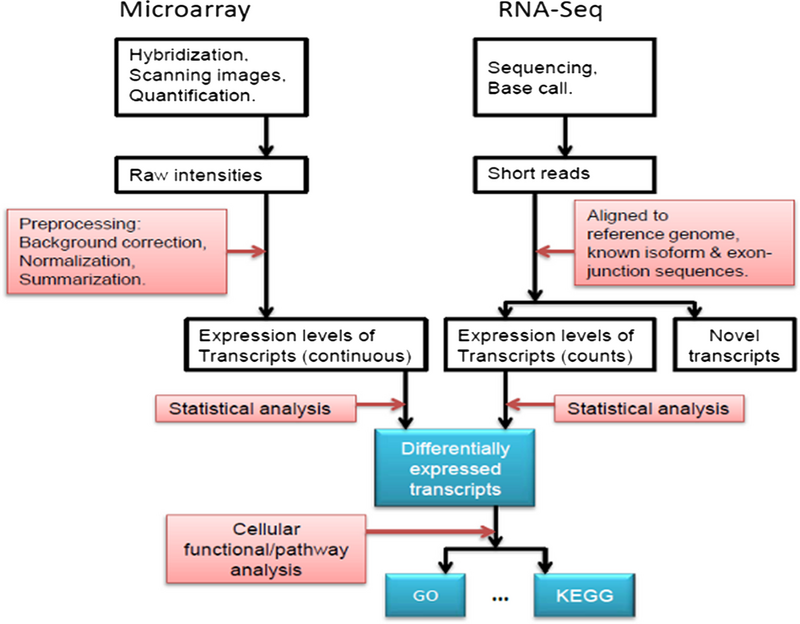 Microarreglo vs RNA-Seq
Microarreglo vs RNA-Seq
Análisis de la expresión de proteínas
Los microarrays de proteínas y la espectrometría de masas (MS) de alto rendimiento (HT) pueden proporcionar una instantánea de las proteínas presentes en una muestra biológica. La bioinformática está muy involucrada en dar sentido a los datos de microarrays de proteínas y HT MS; el primer enfoque se enfrenta a problemas similares a los de los microarreglos dirigidos al ARNm, el último implica el problema de comparar grandes cantidades de datos de masa con las masas predichas de las bases de datos de secuencias de proteínas, y el complicado análisis estadístico de las muestras en las que se detectan múltiples pero incompletos péptidos de cada proteína. detectado. La localización de proteínas celulares en un contexto de tejido se puede lograr a través de la proteómica de afinidad que se muestra como datos espaciales basados en inmunohistoquímica y micromatrices de tejidos.
Análisis de regulación
La regulación génica es la orquestación compleja de eventos mediante los cuales una señal, potencialmente una señal extracelular como una hormona, eventualmente conduce a un aumento o disminución en la actividad de una o más proteínas. Se han aplicado técnicas bioinformáticas para explorar varios pasos en este proceso.
Por ejemplo, la expresión génica puede ser regulada por elementos cercanos en el genoma. El análisis de promotor implica la identificación y el estudio de motivos de secuencia en el ADN que rodea la región codificante de un gen. Estos motivos influyen en la medida en que esa región se transcribe en ARNm. Los elementos potenciadores alejados del promotor también pueden regular la expresión génica, a través de interacciones de bucles tridimensionales. Estas interacciones pueden determinarse mediante análisis bioinformáticos de experimentos de captura de conformación cromosómica.
Los datos de expresión se pueden utilizar para inferir la regulación de genes: uno podría comparar datos de micromatrices de una amplia variedad de estados de un organismo para formular hipótesis sobre los genes involucrados en cada estado. En un organismo unicelular, uno podría comparar etapas del ciclo celular, junto con varias condiciones de estrés (golpe de calor, inanición, etc.). Luego, se pueden aplicar algoritmos de agrupamiento a esos datos de expresión para determinar qué genes se expresan conjuntamente. Por ejemplo, las regiones aguas arriba (promotores) de genes coexpresados pueden buscarse elementos reguladores sobrerrepresentados. Ejemplos de algoritmos de agrupamiento aplicados en el agrupamiento de genes son el agrupamiento de k-medias, los mapas autoorganizados (SOM), el agrupamiento jerárquico y los métodos de agrupamiento de consenso.
Análisis de la organización celular
Se han desarrollado varios enfoques para analizar la ubicación de orgánulos, genes, proteínas y otros componentes dentro de las células. Esto es relevante ya que la ubicación de estos componentes afecta los eventos dentro de una célula y, por lo tanto, nos ayuda a predecir el comportamiento de los sistemas biológicos. Se ha ideado una categoría de ontología génica, componente celular, para capturar la localización subcelular en muchas bases de datos biológicas.
Microscopía y análisis de imágenes
Las imágenes microscópicas nos permiten localizar tanto orgánulos como moléculas. También puede ayudarnos a distinguir entre células normales y anormales, por ejemplo, en el cáncer.
Localización de proteínas
La localización de proteínas nos ayuda a evaluar el papel de una proteína. Por ejemplo, si se encuentra una proteína en el núcleo, puede estar involucrada en la regulación o empalme de genes. Por el contrario, si una proteína se encuentra en las mitocondrias, puede estar involucrada en la respiración u otros procesos metabólicos. La localización de proteínas es, por lo tanto, un componente importante de la predicción de la función de las proteínas. Hay recursos de predicción de localización subcelular de proteínas bien desarrollados disponibles, incluidas bases de datos de localización subcelular de proteínas y herramientas de predicción.
Organización nuclear de la cromatina
Los datos de experimentos de captura de conformación cromosómica de alto rendimiento, como Hi-C (experimento) y ChIA-PET, pueden proporcionar información sobre la proximidad espacial de los loci de ADN. El análisis de estos experimentos puede determinar la estructura tridimensional y la organización nuclear de la cromatina. Los desafíos bioinformáticos en este campo incluyen la partición del genoma en dominios, como los dominios de asociación topológica (TAD), que se organizan juntos en un espacio tridimensional.
Bioinformática estructural
La predicción de la estructura de proteínas es otra aplicación importante de la bioinformática. La secuencia de aminoácidos de una proteína, la denominada estructura primaria, se puede determinar fácilmente a partir de la secuencia del gen que la codifica. En la gran mayoría de los casos, esta estructura primaria determina de manera única una estructura en su entorno nativo. (Por supuesto, hay excepciones, como el prión de la encefalopatía espongiforme bovina (enfermedad de las vacas locas).) El conocimiento de esta estructura es vital para comprender la función de la proteína. La información estructural generalmente se clasifica como secundaria, terciaria y cuaternaria.estructura. Una solución general viable para tales predicciones sigue siendo un problema abierto. La mayoría de los esfuerzos hasta ahora se han dirigido hacia heurísticas que funcionan la mayor parte del tiempo.
Una de las ideas clave en bioinformática es la noción de homología. En la rama genómica de la bioinformática, la homología se utiliza para predecir la función de un gen: si la secuencia del gen A, cuya función se conoce, es homóloga a la secuencia del gen B, cuya función se desconoce, se podría inferir que B puede comparte la función de A. En la rama estructural de la bioinformática, la homología se utiliza para determinar qué partes de una proteína son importantes en la formación de estructuras y la interacción con otras proteínas. En una técnica llamada modelado de homología, esta información se utiliza para predecir la estructura de una proteína una vez que se conoce la estructura de una proteína homóloga. Actualmente, esta sigue siendo la única forma de predecir estructuras de proteínas de manera confiable.
Un ejemplo de esto es la hemoglobina humana y la hemoglobina de las legumbres (leghemoglobina), que son parientes lejanos de la misma superfamilia de proteínas. Ambos cumplen el mismo propósito de transportar oxígeno en el organismo. Aunque ambas proteínas tienen secuencias de aminoácidos completamente diferentes, sus estructuras proteicas son prácticamente idénticas, lo que refleja sus propósitos casi idénticos y su ancestro compartido.
Otras técnicas para predecir la estructura de las proteínas incluyen el enhebrado de proteínas y el modelado basado en la física de novo (desde cero).
Otro aspecto de la bioinformática estructural incluye el uso de estructuras de proteínas para modelos de cribado virtual, como los modelos de relación estructura-actividad cuantitativa y los modelos proteoquimiométricos (PCM). Además, la estructura cristalina de una proteína se puede utilizar en la simulación de, por ejemplo, estudios de unión a ligandos y estudios de mutagénesis in silico.
Biología de redes y sistemas
El análisis de redes busca comprender las relaciones dentro de las redes biológicas, como las redes metabólicas o de interacción proteína-proteína. Aunque las redes biológicas se pueden construir a partir de un solo tipo de molécula o entidad (como los genes), la biología de redes a menudo intenta integrar muchos tipos de datos diferentes, como proteínas, moléculas pequeñas, datos de expresión génica y otros, que están todos conectados físicamente., funcionalmente, o ambos.
La biología de sistemas involucra el uso de simulaciones por computadora de subsistemas celulares (como las redes de metabolitos y enzimas que comprenden el metabolismo, las vías de transducción de señales y las redes reguladoras de genes) para analizar y visualizar las conexiones complejas de estos procesos celulares. La vida artificial o evolución virtual intenta comprender los procesos evolutivos a través de la simulación por computadora de formas de vida simples (artificiales).
Redes de interacción molecular
Se han determinado decenas de miles de estructuras de proteínas tridimensionales mediante cristalografía de rayos X y espectroscopia de resonancia magnética nuclear de proteínas (RMN de proteínas) y una pregunta central en bioinformática estructural es si es práctico predecir posibles interacciones proteína-proteína solo en base a estos Formas 3D, sin realizar experimentos de interacción proteína-proteína. Se han desarrollado una variedad de métodos para abordar el problema del acoplamiento proteína-proteína, aunque parece que aún queda mucho trabajo por hacer en este campo.
Otras interacciones encontradas en el campo incluyen proteína-ligando (incluido el fármaco) y proteína-péptido. La simulación dinámica molecular del movimiento de los átomos sobre enlaces giratorios es el principio fundamental detrás de los algoritmos computacionales, denominados algoritmos de acoplamiento, para estudiar las interacciones moleculares.
Otros
Análisis de la literatura
El crecimiento en la cantidad de literatura publicada hace que sea prácticamente imposible leer todos los artículos, lo que da como resultado subcampos de investigación inconexos. El análisis de la literatura tiene como objetivo emplear la lingüística computacional y estadística para explotar esta creciente biblioteca de recursos de texto. Por ejemplo:
- Reconocimiento de abreviaturas: identifique la forma larga y la abreviatura de términos biológicos
- Reconocimiento de entidades nombradas: reconocimiento de términos biológicos como nombres de genes
- Interacción proteína-proteína: identifique qué proteínas interactúan con qué proteínas del texto
El área de investigación se basa en la estadística y la lingüística computacional.
Análisis de imágenes de alto rendimiento
Las tecnologías computacionales se utilizan para acelerar o automatizar por completo el procesamiento, la cuantificación y el análisis de grandes cantidades de imágenes biomédicas con alto contenido de información. Los sistemas modernos de análisis de imágenes aumentan la capacidad de un observador para realizar mediciones a partir de un conjunto de imágenes grande o complejo, al mejorar la precisión, la objetividad o la velocidad. Un sistema de análisis completamente desarrollado puede reemplazar completamente al observador. Aunque estos sistemas no son exclusivos de las imágenes biomédicas, las imágenes biomédicas se están volviendo más importantes tanto para el diagnóstico como para la investigación. Algunos ejemplos son:
- cuantificación de alto rendimiento y alta fidelidad y localización subcelular (detección de alto contenido, citohistopatología, informática de bioimagen)
- morfometría
- análisis y visualización de imágenes clínicas
- determinar los patrones de flujo de aire en tiempo real en los pulmones de animales vivos
- cuantificar el tamaño de la oclusión en imágenes en tiempo real a partir del desarrollo y la recuperación durante una lesión arterial
- hacer observaciones de comportamiento a partir de grabaciones de video extendidas de animales de laboratorio
- mediciones infrarrojas para la determinación de la actividad metabólica
- inferir superposiciones de clones en el mapeo de ADN, por ejemplo, la puntuación de Sulston
Análisis de datos unicelulares de alto rendimiento
Las técnicas computacionales se utilizan para analizar datos de una sola célula de alto rendimiento y baja medición, como los obtenidos de la citometría de flujo. Estos métodos generalmente implican encontrar poblaciones de células que son relevantes para un estado de enfermedad o condición experimental particular.
Informática de la biodiversidad
La informática de la biodiversidad se ocupa de la recopilación y el análisis de datos de biodiversidad, como bases de datos taxonómicas o datos de microbiomas. Los ejemplos de tales análisis incluyen filogenética, modelado de nichos, mapeo de riqueza de especies, códigos de barras de ADN o herramientas de identificación de especies.
Ontologías e integración de datos
Las ontologías biológicas son grafos acíclicos dirigidos de vocabularios controlados. Están diseñados para capturar conceptos y descripciones biológicas de una manera que se puedan categorizar y analizar fácilmente con computadoras. Cuando se categoriza de esta manera, es posible obtener valor agregado del análisis holístico e integrado.
La OBO Foundry fue un esfuerzo por estandarizar ciertas ontologías. Una de las más difundidas es la ontología génica que describe la función de los genes. También hay ontologías que describen fenotipos.
Bases de datos
Las bases de datos son esenciales para la investigación y las aplicaciones de la bioinformática. Existen muchas bases de datos que cubren varios tipos de información: por ejemplo, secuencias de ADN y proteínas, estructuras moleculares, fenotipos y biodiversidad. Las bases de datos pueden contener datos empíricos (obtenidos directamente de experimentos), datos predichos (obtenidos de análisis) o, más comúnmente, ambos. Pueden ser específicos de un organismo, ruta o molécula de interés en particular. Alternativamente, pueden incorporar datos compilados de muchas otras bases de datos. Estas bases de datos varían en su formato, mecanismo de acceso y si son públicas o no.
Algunas de las bases de datos más utilizadas se enumeran a continuación. Para obtener una lista más completa, consulte el enlace al comienzo de la subsección.
- Utilizado en análisis de secuencias biológicas: Genbank, UniProt
- Utilizado en el análisis de estructuras: Protein Data Bank (PDB)
- Utilizado en la búsqueda de familias de proteínas y búsqueda de motivos: InterPro, Pfam
- Utilizado para la secuenciación de próxima generación: archivo de lectura de secuencias
- Utilizado en análisis de red: bases de datos de vías metabólicas (KEGG, BioCyc), bases de datos de análisis de interacción, redes funcionales
- Utilizado en diseño de circuitos genéticos sintéticos: GenoCAD
Software y herramientas
Las herramientas de software para la bioinformática van desde simples herramientas de línea de comandos hasta programas gráficos más complejos y servicios web independientes disponibles en varias empresas de bioinformática o instituciones públicas.
Software de bioinformática de código abierto
Muchas herramientas de software gratuitas y de código abierto han existido y han seguido creciendo desde la década de 1980. La combinación de una necesidad continua de nuevos algoritmos para el análisis de tipos emergentes de lecturas biológicas, el potencial para experimentos in silico innovadores y bases de código abierto disponibles gratuitamente han ayudado a crear oportunidades para que todos los grupos de investigación contribuyan tanto a la bioinformática como a la gama. de software de código abierto disponible, independientemente de sus acuerdos de financiación. Las herramientas de código abierto a menudo actúan como incubadoras de ideas o complementos respaldados por la comunidad en aplicaciones comerciales. También pueden proporcionar estándares de facto y modelos de objetos compartidos para ayudar con el desafío de la integración de la bioinformación.
La gama de paquetes de software de código abierto incluye títulos como Bioconductor, BioPerl, Biopython, BioJava, BioJS, BioRuby, Bioclipse, EMBOSS,.NET Bio, Orange con su complemento de bioinformática, Apache Taverna, UGENE y GenoCAD. Para mantener esta tradición y crear más oportunidades, la Open Bioinformatics Foundation sin fines de lucro ha apoyado la Conferencia anual de código abierto de bioinformática (BOSC) desde 2000.
Un método alternativo para crear bases de datos bioinformáticas públicas es utilizar el motor MediaWiki con la extensión WikiOpener. Este sistema permite que todos los expertos en la materia accedan y actualicen la base de datos.
Servicios web en bioinformática
Se han desarrollado interfaces basadas en SOAP y REST para una amplia variedad de aplicaciones bioinformáticas que permiten que una aplicación que se ejecuta en una computadora en una parte del mundo use algoritmos, datos y recursos informáticos en servidores en otras partes del mundo. Las principales ventajas se derivan del hecho de que los usuarios finales no tienen que lidiar con los gastos generales de mantenimiento de software y bases de datos.
El EBI clasifica los servicios básicos de bioinformática en tres categorías: SSS (Servicios de búsqueda de secuencias), MSA (Alineación de secuencias múltiples) y BSA (Análisis de secuencias biológicas). La disponibilidad de estos recursos bioinformáticos orientados a servicios demuestra la aplicabilidad de las soluciones bioinformáticas basadas en la web, y van desde una colección de herramientas independientes con un formato de datos común bajo una interfaz única, independiente o basada en la web, hasta bioinformática integradora, distribuida y extensible. sistemas de gestión de flujo de trabajo.
Sistemas de gestión de flujo de trabajo de bioinformática
Un sistema de gestión de flujo de trabajo de bioinformática es una forma especializada de un sistema de gestión de flujo de trabajo diseñado específicamente para componer y ejecutar una serie de pasos computacionales o de manipulación de datos, o un flujo de trabajo, en una aplicación de bioinformática. Tales sistemas están diseñados para
- proporcionar un entorno fácil de usar para que los científicos de aplicaciones individuales creen sus propios flujos de trabajo,
- proporcionar herramientas interactivas para los científicos que les permitan ejecutar sus flujos de trabajo y ver sus resultados en tiempo real,
- simplificar el proceso de compartir y reutilizar flujos de trabajo entre los científicos, y
- permita a los científicos rastrear la procedencia de los resultados de la ejecución del flujo de trabajo y los pasos de creación del flujo de trabajo.
Algunas de las plataformas que dan este servicio: Galaxy, Kepler, Taverna, UGENE, Anduril, HIVE.
Objetos BioCompute y BioCompute
En 2014, la Administración de Drogas y Alimentos de EE. UU. patrocinó una conferencia celebrada en el campus de Bethesda de los Institutos Nacionales de Salud para discutir la reproducibilidad en bioinformática. Durante los siguientes tres años, un consorcio de partes interesadas se reunió regularmente para discutir lo que se convertiría en el paradigma BioCompute. Estas partes interesadas incluyeron representantes del gobierno, la industria y entidades académicas. Los líderes de la sesión representaron numerosas sucursales de los Institutos y Centros de la FDA y los NIH, entidades sin fines de lucro, como el Proyecto Varioma Humano y la Federación Europea de Informática Médica, e instituciones de investigación, como Stanford, el Centro del Genoma de Nueva York y la Universidad George Washington.
Se decidió que el paradigma BioCompute tendría la forma de 'cuadernos de laboratorio' digitales que permiten la reproducibilidad, replicación, revisión y reutilización de protocolos bioinformáticos. Esto se propuso para permitir una mayor continuidad dentro de un grupo de investigación en el curso del flujo normal de personal mientras se fomenta el intercambio de ideas entre grupos. La FDA de EE. UU. financió este trabajo para que la información sobre los oleoductos fuera más transparente y accesible para su personal regulador.
En 2016, el grupo volvió a reunirse en el NIH en Bethesda y discutió el potencial de un objeto BioCompute, una instancia del paradigma BioCompute. Este trabajo se copió como documento de "uso de prueba estándar" y como documento de preimpresión cargado en bioRxiv. El objeto BioCompute permite que el registro en formato JSON se comparta entre empleados, colaboradores y reguladores.
Plataformas educativas
Las plataformas de software diseñadas para enseñar conceptos y métodos de bioinformática incluyen Rosalind y los cursos en línea que se ofrecen a través del portal de capacitación del Instituto Suizo de Bioinformática. Los Talleres Canadienses de Bioinformática ofrecen videos y diapositivas de talleres de capacitación en su sitio web bajo una licencia Creative Commons. El proyecto 4273π o el proyecto 4273pi también ofrece materiales educativos de código abierto de forma gratuita. El curso se ejecuta en computadoras Raspberry Pi de bajo costo y se ha utilizado para enseñar a adultos y escolares. 4273π está desarrollado activamente por un consorcio de académicos y personal de investigación que han ejecutado bioinformática de nivel de investigación utilizando computadoras Raspberry Pi y el sistema operativo 4273π.
Las plataformas MOOC también brindan certificaciones en línea en bioinformática y disciplinas relacionadas, incluida la especialización en bioinformática de Coursera (UC San Diego) y la especialización en ciencia de datos genómicos (Johns Hopkins), así como el análisis de datos para Life Sciences XSeries de EdX (Harvard). La Universidad del Sur de California ofrece una Maestría en Bioinformática Traslacional que se enfoca en aplicaciones biomédicas.
Conferencias
Hay varias conferencias importantes que se ocupan de la bioinformática. Algunos de los ejemplos más notables son los Sistemas Inteligentes para Biología Molecular (ISMB), la Conferencia Europea sobre Biología Computacional (ECCB) y la Investigación en Biología Molecular Computacional (RECOMB).
Contenido relacionado
Planta anual
Lista de jardines botánicos
Hierba azul