
Ataxia espinocerebelosa
ataxia espinocerebelosa (SCA) es una enfermedad genética progresiva y degenerativa con múltiples tipos, cada uno de los cuales podría considerarse una afección neurológica por derecho propio. Se estima que 150.000 personas en los Estados Unidos tienen un diagnóstico de ataxia espinocerebelosa en un momento dado. El SCA es hereditario, progresivo, degenerativo y, a menudo, mortal. No se conoce ningún tratamiento o cura eficaz. El SCA puede afectar a cualquier persona de cualquier edad. La enfermedad es causada por un gen recesivo o dominante. En muchos casos, las personas no son conscientes de que son portadoras de un gen relevante hasta que tienen hijos que empiezan a mostrar signos de padecer el trastorno.
Signos y síntomas
La ataxia espinocerebelosa (SCA) pertenece a un grupo de trastornos genéticos caracterizados por una falta de coordinación de la marcha lentamente progresiva y, a menudo, se asocia con una mala coordinación de las manos, el habla y los movimientos oculares. Recientemente se publicó una revisión de diferentes características clínicas entre los subtipos de SCA, describiendo la frecuencia de características no cerebelosas, como parkinsonismo, corea, piramidalismo, deterioro cognitivo, neuropatía periférica, convulsiones, entre otras. Al igual que con otras formas de ataxia, el SCA frecuentemente produce atrofia del cerebelo, pérdida de la coordinación fina de los movimientos musculares que conduce a movimientos inestables y torpes, y otros síntomas. Los déficits oculares se pueden cuantificar mediante la escala SODA.
Los síntomas de una ataxia varían según el tipo específico y el paciente individual. En muchos casos una persona con ataxia conserva plena capacidad mental pero pierde progresivamente el control físico.
Causa
Las ataxias hereditarias se clasifican por modo de herencia y gen causante o locus cromosómico. Las ataxias hereditarias pueden heredarse de forma autosómica dominante, autosómica recesiva o ligada al cromosoma X.
- Se conocen muchos tipos de ataxias cerebelosas autosómicas dominantes para las que se dispone de información genética específica. Los sinónimos de ataxias cerebelosas autosómicas (ADCA) utilizados antes de la comprensión actual de la genética molecular fueron la ataxia de Marie, la atrofia olivopontocerebelosa heredada, la atrofia cerebello-olivaria o el término más genérico "degeneración espinocerebelosa". ()Degeneración Spinocerebellar es un raro trastorno neurológico hereditario del sistema nervioso central caracterizado por la lenta degeneración de ciertas áreas del cerebro. Hay tres formas de degeneración spinocerebellar: Tipos 1, 2, 3. Los síntomas comienzan durante la edad adulta.)
- Hay cinco típicos autosómico-recesivo trastornos en los que la ataxia es una característica prominente: Friedreich ataxia, ataxia-telangiectasia, ataxia con deficiencia de vitamina E, ataxia con apraxia oculomotor (AOA), ataxia espástica. Subdivisiones desordenadas: ataxia de Friedreich, ataxia spinocerebellar, ataxia telangiectasia, vasomotor ataxia, vestibulocerebellar, ataxiadynamia, ataxiofemia y atrofia olivopontocerebellar.
- Se ha informado de casos en que una expansión de poliglutamina puede alargarse cuando se transmite, lo que a menudo puede dar lugar a una edad temprana y a un fenotipo de enfermedad más grave para las personas que heredan el alelo de la enfermedad. Esto cae bajo la categoría de anticipación genética. Varios tipos de SCA se caracterizan por la expansión repetitiva de la secuencia de trinucleótidos CAG en ADN que codifica un tracto repetitivo de poliglutamina en proteínas. La expansión de CAG repite a lo largo de las generaciones sucesivas parece deberse a la falta de pauta deslizada durante la replicación del ADN o la reparación del ADN.
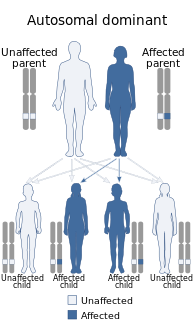 Hay numerosos tipos de ataxias cerebelosas autosómicas
Hay numerosos tipos de ataxias cerebelosas autosómicas Hay cinco trastornos típicos autosómicos recesivos en los que la ataxia es una característica prominente
Hay cinco trastornos típicos autosómicos recesivos en los que la ataxia es una característica prominente


Diagnóstico
Clasificación
Unos pocos SCA siguen sin especificar y no pueden ser diagnosticados precisamente, pero en la última década las pruebas genéticas han permitido la identificación precisa de docenas de diferentes SCA y se están agregando más pruebas cada año. En 2008, se desarrolló un análisis de sangre de ataxia genética para probar 12 tipos de SCA, la ataxia de Friedreich y varios otros. Sin embargo, dado que no todos los SCA han sido identificados genéticamente, algunos SCA todavía se diagnostican mediante exámenes neurológicos, que pueden incluir un examen físico, antecedentes familiares, análisis de resonancia magnética del cerebro y la columna vertebral, y el grifo espinal.
Muchas de las SCA siguientes se incluyen en la categoría de enfermedades por poliglutamina, que se producen cuando una proteína asociada a una enfermedad (es decir, ataxina-1, ataxina-3, etc.) contiene una gran cantidad de repeticiones de residuos de glutamina, denominada poliQ. secuencia o una "repetición de trinucleótido CAG" enfermedad para la designación de una letra o el codón para glutamina, respectivamente. El umbral para los síntomas en la mayoría de las formas de SCA es de alrededor de 35, aunque para SCA3 se extiende más allá de 50. La mayoría de las enfermedades por poliglutamina son dominantes debido a las interacciones de la cola poliQ resultante.
El primer gen de la ataxia se identificó en 1993 y se denominó "ataxia espinocerebelosa tipo 1" (SCA1); los genes posteriores se denominaron SCA2, SCA3, etc. Por lo general, el "tipo" número de "SCA" Se refiere al orden en que se encontró el gen. En este momento, se han encontrado al menos 49 mutaciones genéticas diferentes.
Lo siguiente es una lista de algunos de los muchos tipos de Ataxia Spinocerebellar.
| SCA Tipo | Activo medio (Range in Years) | Duración media (Range in Years) | Lo que el paciente experimenta | Origen común | Problemas con ADN |
|---|---|---|---|---|---|
| SCA1 (ATXN1) | 4a década ( 10 a 60) | 15 años (10 a 35) | Sacadas hipermétricas, saccaderías lentas, neurona motora superior (nota: saccades se relaciona con el movimiento ocular) | CAG repetir, 6p (Ataxin 1) | |
| SCA2 (ATXN2) | Tercera y cuarta década ( 10 a 60) | 10 años (1–30) | Sacadas de velocidad reducida areflexia (absencia de reflejos neurológicos) | Cuba | CAG repetir, 12q |
| SCA3 (MJD) (ATXN3) | 4a década (10 a 70) | 10 años (1–20) | También se llama enfermedad de Machado-Joseph (MJD) Nistagmus (un movimiento rápido, involuntario y oscilatorio del globo ocular) neurona motora superior lentas saccades | Azores (Portugal) | CAG repetir, 14q |
| SCA4 (PLEKHG4) | 4a y séptima década (19–72) | Decenios | areflexia (absencia de reflejos neurológicos) | Cromosoma 16q | |
| SCA5 (SPTBN2) | Tercera y cuarta década (10 a 68) | 25 años | Cerebello puro | Cromosoma 11 | |
| SCA6 (CACNA1A) | 5a y 6a década (19–71) | 25 años | Nistagmus, vértigo posicional Los síntomas pueden aparecer por primera vez tan tarde como 65 años. | CAG repetir, 19p gen de canal de calcio | |
| SCA7 (ATXN7) | Tercera y cuarta década (0,5-60) | 20 años (1–45; el inicio temprano correlaciona con menor duración) | Degeneración macular, neurona motor superior, lentas saccades | CAG repetir, 3p (Ataxin 7) | |
| SCA8 (IOSCA) | 39 años (18–65) | Vidas normales | Nistagmo horizontal (un movimiento rápido, involuntario y oscilatorio del globo ocular), inestabilidad, falta de coordinación | Repetición del CTG, 13q | |
| SCA10 (ATXN10) | 36 años | 9 años | ataxia, convulsiones | México | Cromosoma 22q unido pentanucleótido repetir |
| SCA11 (TTBK2) | 30 años (15 a 70) | Vidas normales | Mild, permanece ambulatorio (puede caminar por uno mismo) | 15q | |
| SCA12 (PPP2R2B) | 33 años (8 a 55) | Temblor de cabeza y mano, akinesia (pérdida de la función motor normal, resultando en el movimiento muscular deficiente) | CAG repetir, 5q | ||
| SCA13 (KCNC3) | Niñez o edad adulta dependiendo de la mutación | Según KCNC3 (una especie de gen) | Discapacidad intelectual | 19q | |
| (PRKCG) | 28 años (12–42) | Decenios (1–30) | Myoclonus (un retoque repentino de los músculos o partes de los músculos, sin ningún ritmo o patrón, que ocurre en varios trastornos cerebrales) | 19q | |
| SCA16 (ITPR1) | 39 años (20 a 66) | 1 a 40 años | Temblor de cabeza y mano | 8q | |
| SCA17 (TBP) | CAG repetir, 6q (proteína de unión de TTA) | ||||
| SCA19, SCA22 (KCND3) | Síndrome de cerebella leve, disarthria | ||||
| SCA25 | 1,5 a 39 años | Desconocido | ataxia con neuropatía sensorial, vómitos y dolor gastrointestinal. | 2p | |
| SCA27 (FGF14) | 15 a 20 años | Desconocido | ataxia con pobre cognición, disquinesias y temblor. | FGF14 13q34 | |
| SCA35 | 40 a 48 años | Desconocido | gait and limb ataxia, disarthria, dismetria ocular, temblor de intención, pseudobulbar palsy, torticollis spasmodic, extensor plantar respuestas, propriocepción reducida e hiperreflexia | China | transglutaminase 6 (TGM6) localizado en cromosoma 20p13 |
| SCA36 | Quinto y sexto decenio (adulto) | Decenios | ataxia, hiperrheflexia, disarthria, fasciculaciones de la lengua con posterior desperdicio de la lengua | NOP56 | |
| SCA37 | Adulto | Decenios | disarthria, gait lentamente progresivo y ataxia de miembros con dismetria severa en las extremidades inferiores, dismetria suave en las extremidades superiores, disfagia y movimientos oculares anormales | DAB1 |
Otros incluyen SCA18, SCA20, SCA21, SCA23, SCA26, SCA28 y SCA29.
Se han descrito cuatro tipos conectados con X (302500, 302600, 301790, 301840), pero sólo el primero de ellos se ha vinculado hasta ahora a un gen (SCAX1).
| Nombre | OMIM | Enfermedades raras | Otros |
|---|---|---|---|
| Anemia, ataxia de spinocerebelal sideroblástica; síndrome de Detter de aves de pago | 301310 | ID de enfermedad 668 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia de Friedreich; ataxia de Spinocerebellar, Friedreich | 229300 | ID de enfermedad 6468 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia de Spinocerebellar infantil | 605361 | ID de enfermedad 4062 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 1 | 164400 | ID de enfermedad 4071 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 2 | 183090 | ID de enfermedad 4072 en la Oficina de Enfermedades Raras de NIH | |
| Enfermedad de Machado Joseph | 109150 | ID de enfermedad 6801 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 4 | 600223 | ID de enfermedad 9970 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 5 | 600224 | ID de enfermedad 4953 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 7 | 164500 | ID de enfermedad 4955 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 8 | 603680 | ID de enfermedad 4956 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 13 | 605259 | ID de enfermedad 9611 en la Oficina de Enfermedades Raras de NIH | |
| Spinocerebellar ataxia 18 | 607458 | ID de enfermedad 9976 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 19 | 607346 | ID de enfermedad 9969 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 20 | 608687 | ID de enfermedad 9997 en la Oficina de Enfermedades Raras de NIH | |
| Spinocerebellar ataxia 21 | 607454 | ID de enfermedad 9999 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 23 | 610245 | ID de enfermedad 9950 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 25 | 608703 | ID de enfermedad 9996 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 26 | 609306 | ID de enfermedad 9995 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 28 | 610246 | ID de enfermedad 9951 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 30 | 117360 | ID de enfermedad 9975 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar 35 | 613908 | ID de enfermedad en la Oficina de Enfermedades Raras de NIH | |
| Síndrome de sordera de ataxia espinacerebelosa | ID de enfermedad 2451 en la Oficina de Enfermedades Raras de NIH | ORPHA:2074 en Orphanet | |
| Ataxia Spinocerebellar, recesivo autosómico 1 | 606002 | ID de enfermedad 4949 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, recesivo autosómico 3 | 271250 | ID de enfermedad 9971 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, recesivo autosómico 4 | 607317 | ID de enfermedad 4952 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, recesivo autosómico 5 | 606937 | ID de enfermedad 9977 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, recesivo autosómico 6 | 608029 | ID de enfermedad 4954 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, recesivo autosómico 21 - mutación en SCYL1 | Herencia Mendeliana en línea en el hombre (OMIM): 616719 | ORPHA:466794 | |
| Ataxia Spinocerebellar, recesivo autosómico, con neuropatía axonal | 607250 | ID de enfermedad 10000 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, X-linked, 2 | 302600 | ID de enfermedad 9978 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, X-linked, 3 | 301790 | ID de enfermedad 9981 en la Oficina de Enfermedades Raras de NIH | |
| Ataxia Spinocerebellar, X-linked, 4 | 301840 | ID de enfermedad 9980 en la Oficina de Enfermedades Raras de NIH |
Tratamiento
Medicamento
No existe cura para la ataxia espinocerebelosa, que actualmente se considera una enfermedad progresiva e irreversible, aunque no todos los tipos causan una discapacidad igualmente grave.
En general, los tratamientos están dirigidos a aliviar los síntomas, no la enfermedad en sí. Muchos pacientes con formas hereditarias o idiopáticas de ataxia tienen otros síntomas además de la ataxia. Los medicamentos u otras terapias pueden ser apropiados para algunos de estos síntomas, que pueden incluir temblores, rigidez, depresión, espasticidad y trastornos del sueño, entre otros. Tanto la aparición de los síntomas iniciales como la duración de la enfermedad son variables. Si la enfermedad es causada por una expansión CAG repetida del trinucleótido de poliglutamina, una expansión más prolongada puede conducir a un inicio más temprano y una progresión más radical de los síntomas clínicos. Por lo general, una persona con esta enfermedad eventualmente no podrá realizar las tareas diarias (AVD). Sin embargo, los terapeutas de rehabilitación pueden ayudar a los pacientes a maximizar su capacidad de autocuidado y retrasar el deterioro hasta cierto punto. Los investigadores están explorando múltiples vías para encontrar una cura, incluida la interferencia del ARN (ARNi), el uso de células madre y varias otras vías.
El 18 de enero de 2017, BioBlast Pharma anunció la finalización de los ensayos clínicos de fase 2a de su medicamento, trehalosa, en el tratamiento de SCA3. BioBlast ha recibido el estado de vía rápida de la FDA y el estado de medicamento huérfano para su tratamiento. La información proporcionada por BioBlast en su investigación indica que esperan que este tratamiento pueda resultar eficaz en otros tratamientos de SCA que tengan patología similar relacionada con las enfermedades PolyA y PolyQ.
Además, la Dra. Beverly Davidson ha estado trabajando en una metodología que utiliza tecnología RNAi para encontrar una cura potencial durante más de 2 décadas. Su investigación comenzó a mediados de la década de 1990 y avanzó hasta trabajar con modelos de ratones aproximadamente una década después y, más recientemente, pasó a un estudio con primates no humanos. Los resultados de su investigación más reciente "respaldan la aplicación clínica de esta terapia génica".
Finalmente, Boudreau et al. también han demostrado otra tecnología de transferencia de genes descubierta en 2011. parece muy prometedor y ofrece otra vía hacia una posible cura futura.
N-Acetil-Leucina
La N-Acetil-Leucina es un aminoácido modificado administrado por vía oral que IntraBio Inc (Oxford, Reino Unido) está desarrollando como un tratamiento novedoso para múltiples trastornos neurológicos raros y comunes.
La N-Acetil-Leucina ha recibido múltiples designaciones de medicamento huérfano de la U.S. Food & Administración de Medicamentos (FDA) y la Agencia Europea de Medicamentos (EMA) para el tratamiento de diversas enfermedades genéticas, incluidas las ataxias espinocerebelosas. A la N-acetil-leucina también se le han otorgado designaciones de medicamento huérfano en los EE. UU. y la UE para la ataxia cerebelosa hereditaria relacionada, ataxia-telangiectasia. U.S. Food & Administración de Medicamentos (FDA) y la Agencia Europea de Medicamentos (EMA).
Los estudios de series de casos publicados han demostrado los efectos del tratamiento agudo con N-acetil-leucina para el tratamiento de ataxias cerebelosas hereditarias, incluidas las ataxias espinocerebelosas. Estos estudios demostraron además que el tratamiento es bien tolerado y tiene un buen perfil de seguridad. En 2019 comenzó un ensayo clínico multinacional que investiga la N-Acetil-L-Leucina para el tratamiento de una ataxia cerebelosa hereditaria relacionada, la ataxia-telangiectasia.
IntraBio también está realizando ensayos clínicos paralelos con N-Acetil-L-Leucina para el tratamiento de la enfermedad de Niemann-Pick tipo C y la gangliosidosis GM2 (enfermedad de Tay-Sachs y Sandhoff). Las oportunidades futuras para desarrollar N-acetil-leucina incluyen la demencia con cuerpos de Lewy, la esclerosis lateral amiotrófica, el síndrome de piernas inquietas, la esclerosis múltiple y la migraña.
Rehabilitación
Los fisioterapeutas pueden ayudar a los pacientes a mantener su nivel de independencia a través de programas de ejercicio terapéutico. Un informe de investigación reciente demostró una ganancia de dos puntos SARA (Escala para la evaluación y clasificación de la ataxia) con la fisioterapia. En general, la fisioterapia enfatiza el equilibrio postural y el entrenamiento de la marcha para los pacientes con ataxia. El acondicionamiento general, como ejercicios de amplitud de movimiento y fortalecimiento muscular, también se incluiría en los programas de ejercicios terapéuticos. La investigación demostró que los pacientes con ataxia espinocerebelosa 2 (SCA2) con una etapa leve de la enfermedad obtuvieron una mejora significativa en el equilibrio estático y los índices neurológicos después de seis meses de un programa de entrenamiento con ejercicios de fisioterapia. Los terapeutas ocupacionales pueden ayudar a los pacientes con problemas de falta de coordinación o ataxia mediante el uso de dispositivos adaptativos. Dichos dispositivos pueden incluir un bastón, muletas, un andador o una silla de ruedas para personas con problemas para caminar. Hay otros dispositivos disponibles para ayudar con la escritura, la alimentación y el cuidado personal si la coordinación de manos y brazos está afectada. Un ensayo clínico aleatorizado reveló que un programa intensivo de rehabilitación con terapias físicas y ocupacionales para pacientes con enfermedades cerebelosas degenerativas puede mejorar significativamente las ganancias funcionales en la ataxia, la marcha y las actividades de la vida diaria. Se demostró que se mantenía cierto nivel de mejora 24 semanas después del tratamiento. Los logopedas pueden utilizar tanto estrategias de intervención conductual como dispositivos de comunicación aumentativos y alternativos para ayudar a los pacientes con problemas del habla.